一种基于种群拥挤度识别的差分进化蛋白质结构预测方法与流程
本发明涉及生物信息学、计算机应用领域,尤其涉及的是一种基于种群拥挤度识别的差分进化蛋白质结构预测方法。
背景技术:
蛋白质分子在生物细胞化学反应过程中起着至关重要的作用。它们的结构模型和生物活性状态对我们理解和治愈多种疾病有重要的意义。蛋白质只有折叠成特定的三维结构才能产生其特有的生物学功能。因此,要了解蛋白质的功能,就必须获得其三维空间结构。
生物信息学是生命科学和计算机科学交叉领域的一个研究热点。生物信息学研究成果目前已经被广泛应用于基因发现和预测、基因数据的存储管理、数据检索与挖掘、基因表达数据分析、蛋白质结构预测、基因和蛋白质同源关系预测、序列分析与比对等。目前,根据Anfinsen假设,直接从氨基酸序列出发,基于势能模型,采用全局优化方法,搜索分子系统的最小能量状态,从而高通量、廉价地预测肽链的天然构象,已经成为生物信息学最重要的研究课题之一。对于序列相似度低或多肽(<10个残基的小蛋白)来说,从头预测方法是唯一的选择。从头预测方法必须考虑以下两个因素:(1)蛋白质结构能量函数;(2)构象空间搜索方法。第一个因素本质上属于分子力学问题,主要是为了能够计算得到每个蛋白质结构对应的能量值。目前已经存在一些有效的结构能量函数,如简单网格模型HP及更实际的经验力场模型MM3,AMBER,CHARMM,GROMOS,DISCOVER,ECEPP/3等;第二个因素本质上属于全局优化问题,通过选择一种合适的优化方法,对构象空间进行快速搜索,得到与某一全局最小能量对应的构象。其中,蛋白质构象空间优化属于一类非常难解的NP-Hard问题。2005年,D.Baker在Science中指出,构象空间优化方法是制约蛋白质从头预测方法预测精度的一个瓶颈因素。
因此,现有的蛋白质结构预测方法存在采样效率、复杂度及预测精度方面存在不足,需要改进。
技术实现要素:
为了克服现有蛋白质结构预测存在采样效率低、复杂度较高及预测精度较低 的不足,本发明提出一种采样效率和预测精度较高的基于种群拥挤度识别的差分进化蛋白质结构预测方法。
本发明解决其技术问题所采用的技术方案是:
一种基于种群拥挤度识别的差分进化蛋白质结构预测方法,所述预测方法包括以下步骤:
1)给定查询序列信息;
2)初始化:设置群体规模PopSize,变异因子F,交叉概率CR,片段长度为L,能量函数选用Rosetta Score3,种群拥挤度因子PCL,相似因子SF,迭代次数G=0,迭代次数阈值iteration;首先通过对查询序列随机片段组装,生成一个规模为PopSize的初始种群,初始群体为P={xi|i∈I},计算初始种群个体按照Rosetta Score3计算的能量值f(xi),i∈I,其中i为种群个体编号,I为种群个体编号集合,I={1,2,...,PopSize};
3)依次对种群中的每个个体进行以下操作:
3.1)令i=1,其中i∈{1,2,3,…,PopSize};令Ptarget=Pi,其中i为序号,Ptarget表示目标个体;
3.2)随机生成正整数rand1,rand2,rand3∈{1,2,3,......NP},且rand1≠rand2≠rand3≠i;再生成4个随机整数randrange1,randrange2,randrange3,randrange4;其中randrange1≠randrange2,randrange3≠randrange4∈{1,2,…,Length},Length为序列长度;
3.3)针对个体Pj做变异操作,其中:j=rand1;
3.3.1)令a=min(randrange1,randrange2),b=max(randrange1,randrange2),k∈[a,b];令c=min(randrange3,randrange4),d=max(randrange3,randrange4),p∈[c,d];其中min表示取两个数的最小值,max表示取两个数的最大值;
3.3.2)用Prand2上位置a到位置b的片段的氨基酸所对应的二面角phi、psi、omega替换Pj的相同位置所对应的二面角phi、psi、omega;再使用Prand3上位置c到位置d的片段的氨基酸所对应的二面角phi、psi、omega替换Pj上相同位置所对应的二面角phi、psi、omega,再将所得Pj进行片段组装得到测试个体Ptrail;
3.4)针对测试个体Ptrail做交叉操作:
3.4.1)生成随机数rand4,rand5,其中rand4∈(0,1),rand5∈(1,Length);
3.4.2)根据执行交叉过程:若随机数rand4<=CR,个体Ptrail的片段rand5替换为个体Ptarget中对应的片段,否则直接继承个体Ptrail;
3.5)根据Anfinsen提出的天然构象的蛋白质处于热力学最低的能量状态,比较测试个体Ptrail和目标个体Ptarget的能量函数值,选择能量函数值较低的个体从而更新种群;
3.6)i=i+1,如果i<popSize转至3.2);
4)针对种群中每个个体进行相似度识别:
4.1)令i=1;
4.2)创建一个相似度个体统计变量CS=0及拥挤度个体列表CSList,对目标个体xi∈I(i=1,2,···,popSize)与其他个体比较相似度均方根误差RMSD,并将xi存入拥挤度个体列表CSList中;
4.3)若RMSD<=SF,则令CS=CS+1;
4.4)对个体xi进行种群相似度识别判定,若种群拥挤度因子PCL>1/CS,则表示种群拥挤,对拥挤度列表的个体进行变异操作,令i=i+1,如果i<popSize转至4.2);
5)判断是否满足终止条件,若不满足则转至3),若满足终止条件,则输出结果。进一步,所述步骤5)中,对种群中的每一个个体都执行完步骤3)—4)以后,迭代次数G=G+1,终止条件为迭代次数G达到预设迭代次数阈值iteration。
本发明的技术构思为:在差分进化算法(DE)的框架下,首先,对输入的查询序列进行随机片段组装,生成具有多样化折叠类型的初始构象种群;然后,依次选取种群中的个体作为目标个体,随机选取和目标个体不一样的两个个体进行变异、交叉生成测试个体,根据Rosetta Score3能量函数选取能量较低的个体更新种群;之后,依次对种群中每个个体进行种群相似度比较,根据种群拥挤度因子对拥挤的种群进行变异操作提高种群多样性;在多样性种群个体的指导下避免了算法陷入局部最优,提高了采样效率,预测得到精度更高的诱导构象;在种群拥挤 度识别差分进化的指导下,通过不断更新种群获得一系列亚稳态构象。
本发明的有益效果为:采样效率高、预测精度较高。
附图说明
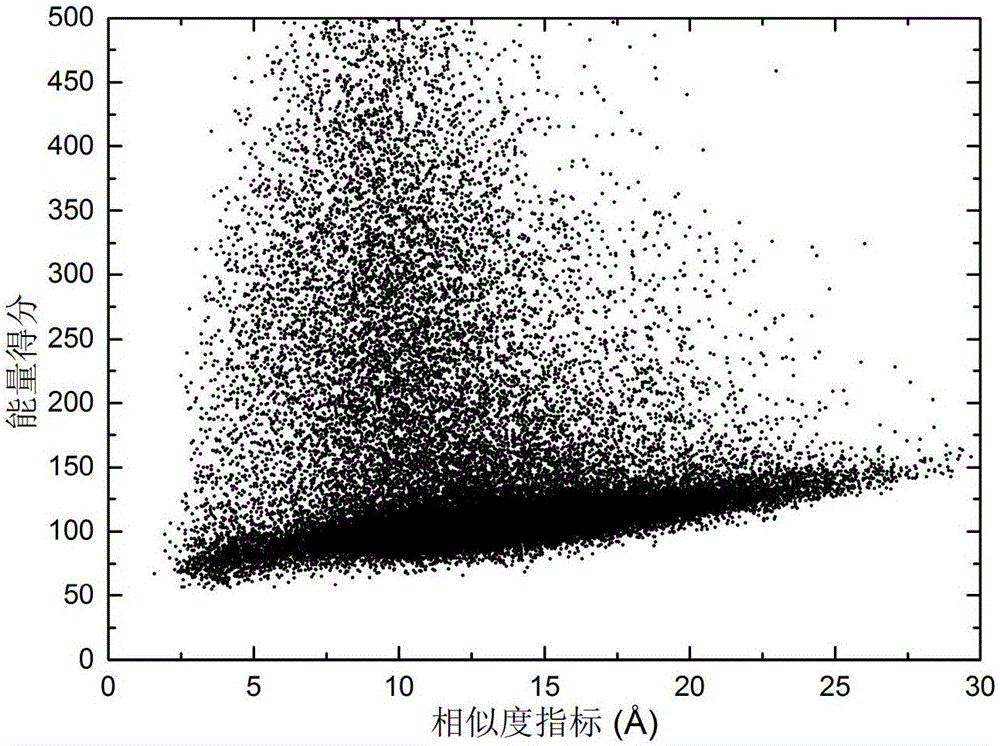
图1是测试序列在种群更新过程中RMSD和能量值的关系示意图。
图2是1AIL在本算法预测所得诱导构象与基本差分进化预测所得构象的采样概率分布图。
具体实施方式
下面结合附图对本发明作进一步描述。
参照图1和图2,一种基于种群拥挤度识别的差分进化蛋白质结构预测方法,包括以下步骤:
1)给定查询序列信息;
2)初始化:设置群体规模PopSize,变异因子F,交叉概率CR,片段长度为L,能量函数选用Rosetta Score3,种群拥挤度因子PCL,相似因子SF,迭代次数G=0,迭代次数阈值iteration;首先通过对查询序列随机片段组装,生成一个规模为PopSize的初始种群,初始群体为P={xi|i∈I},计算初始种群个体按照Rosetta Score3计算的能量值f(xi),i∈I,其中i为种群个体编号,I为种群个体编号集合,I={1,2,...,PopSize};
3)依次对种群中的每个个体进行以下操作:
3.1)令i=1,其中i∈{1,2,3,…,PopSize};令Ptarget=Pi,其中i为序号,Ptarget表示目标个体;
3.2)随机生成正整数rand1,rand2,rand3∈{1,2,3,......NP},且rand1≠rand2≠rand3≠i;再生成4个随机整数randrange1,randrange2,randrange3,randrange4;其中randrange1≠randrange2,randrange3≠randrange4∈{1,2,…,Length},Length为序列长度;
3.3)针对个体Pj做变异操作,其中:j=rand1;
3.3.1)令a=min(randrange1,randrange2),b=max(randrange1,randrange2),k∈[a,b];令c=min(randrange3,randrange4),d=max(randrange3,randrange4),p∈[c,d];其中min表示取两个数的最小值,max表示取两个数的最大值;
3.3.2)用Prand2上位置a到位置b的片段的氨基酸所对应的二面角phi、psi、omega替换Pj的相同位置所对应的二面角phi、psi、omega;再使用Prand3上位置c到位置d的片段的氨基酸所对应的二面角phi、psi、omega替换Pj上相同位置所对应的二面角phi、psi、omega,再将所得Pj进行片段组装得到测试个体Ptrail;
3.4)针对测试个体Ptrail做交叉操作:
3.4.1)生成随机数rand4,rand5,其中rand4∈(0,1),rand5∈(1,Length);
3.4.2)根据执行交叉过程:若随机数rand4<=CR,个体Ptrail的片段rand5替换为个体Ptarget中对应的片段,否则直接继承个体Ptrail;
3.5)根据Anfinsen提出的天然构象的蛋白质处于热力学最低的能量状态,比较测试个体Ptrail和目标个体Ptarget的能量函数值,选择能量函数值较低的个体从而更新种群;
3.6)i=i+1,如果i<popSize转至3.2);
4)针对种群中每个个体进行相似度识别:
4.1)令i=1;
4.2)创建一个相似度个体统计变量CS=0及拥挤度个体列表CSList,对目标个体xi∈I(i=1,2,…,popSize)与其他个体比较相似度均方根误差RMSD,并将xi存入拥挤度个体列表CSList中;
4.3)若RMSD<=SF,则令CS=CS+1;
4.4)对个体xi进行种群相似度识别判定,若种群拥挤度因子PCL>1/CS,则表示种群拥挤,对拥挤度列表的个体进行变异操作,令i=i+1,如果i<popSize转至4.2);
5)判断是否满足终止条件,若不满足则转至3),若满足终止条件,则输出结果。
进一步,所述步骤5)中,对种群中的每一个个体都执行完步骤3)—4)以后,迭代次数G=G+1,终止条件为迭代次数G达到预设迭代次数阈值iteration。
本实施例以序列长度为73的蛋白质1AIL为实施例,一种基于种群拥挤度识别的差分进化蛋白质结构预测方法,其中包含以下步骤:
1)给定查询序列信息;
2)初始化:设置群体规模PopSize=50,变异因子F=0.5,交叉概率CR=0.5,片段长度为L=3,能量函数选用Rosetta Score3,种群拥挤度因子PCL=0.1,相似因子迭代次数G=0,迭代次数阈值为iteration=30000;首先通过对查询序列随机片段组装,生成一个规模为PopSize的初始种群,初始群体为P={xi|i∈I},计算初始种群个体按照Rosetta Score3计算的能量值f(xi),i∈I,其中i为种群个体编号,I为种群个体编号集合,I={1,2,...,PopSize};
3)依次对种群中的每个个体进行以下操作:
3.1)令i=1,其中i∈{1,2,3,…,PopSize};令Ptarget=Pi,其中i为序号,Ptarget表示目标个体;
3.2)随机生成正整数rand1,rand2,rand3∈{1,2,3,......NP},且rand1≠rand2≠rand3≠i;再生成4个随机整数randrange1,randrange2,randrange3,randrange4;其中randrange1≠randrange2,randrange3≠randrange4∈{1,2,…,Length},Length为序列长度;
3.3)针对个体Pj做变异操作,其中:j=rand1;
3.3.1)令a=min(randrange1,randrange2),b=max(randrange1,randrange2),k∈[a,b];令c=min(randrange3,randrange4),d=max(randrange3,randrange4),p∈[c,d];其中min表示取两个数的最小值,max表示取两个数的最大值;
3.3.2)用Prand2上位置a到位置b的片段的氨基酸所对应的二面角phi、psi、omega替换Pj的相同位置所对应的二面角phi、psi、omega;再使用Prand3上位置c到位置d的片段的氨基酸所对应的二面角phi、psi、omega替换Pj上相同位置所对应的二面角phi、psi、omega,再将所得Pj进行片段组装得到测试个体Ptrail;
3.4)针对测试个体Ptrail做交叉操作:
3.4.3)生成随机数rand4,rand5,其中rand4∈(0,1),rand5∈(1,Length);
3.4.4)根据执行交叉过程:若随机数rand4<=CR,个体Ptrail的片段rand5替换为个体Ptarget中 对应的片段,否则直接继承个体Ptrail;
3.5)根据Anfinsen提出的天然构象的蛋白质处于热力学最低的能量状态,比较测试个体Ptrail和目标个体Ptarget的能量函数值,选择能量函数值较低的个体从而更新种群;
3.6)i=i+1,如果i<popSize转至3.2);
4)针对种群中每个个体进行相似度识别:
4.1)令i=1;
4.2)创建一个相似度个体统计变量CS=0及拥挤度个体列表CSList,对目标个体xi∈I(i=1,2,.··,popSize)与其他个体比较相似度均方根误差RMSD,并将xi存入拥挤度个体列表CSList中;
4.3)若RMSD<=SF,则令CS=CS+1;
4.4)对个体xi进行种群相似度识别判定,若种群拥挤度因子PCL>1/CS,则表示种群拥挤,对拥挤度列表的个体进行变异操作,令i=i+1,如果i<popSize转至4.2);
5)对种群中的每一个个体都执行完步骤3)—4)以后,迭代次数G=G+1,终止条件为迭代次数G达到预设迭代次数阈值iteration;判断是否满足终止条件到达迭代次数iteration,若不满足则转至3),若满足终止条件,则输出结果。
以序列长度为73的蛋白质1AIL为实施例,运用以上方法得到了该蛋白质的近天然态构象,构象系综中构象更新图如图1所示,本算法预测所得诱导构象与基本差分进化预测所得构象的采样概率分布图展示如图2所示。
以上阐述的是本发明给出的一个实施例表现出来的优良效果,显然本发明不仅适合上述实施例,在不偏离本发明基本精神及不超出本发明实质内容所涉及内容的前提下可对其做种种变化加以实施。
- 还没有人留言评论。精彩留言会获得点赞!