一种烟用添加剂中二甲苯麝香和芝麻酚的测定方法与流程
本发明属于烟用添加剂的理化检验
技术领域:
,涉及一种烟用添加剂中二甲苯麝香和芝麻酚的测定方法,具体涉及一种对烟用添加剂中二甲苯麝香和芝麻酚,采用同位素稀释-分散液液萃取法进行萃取、富集与净化,并采用气相色谱质谱联用法进行测定的方法。
背景技术:
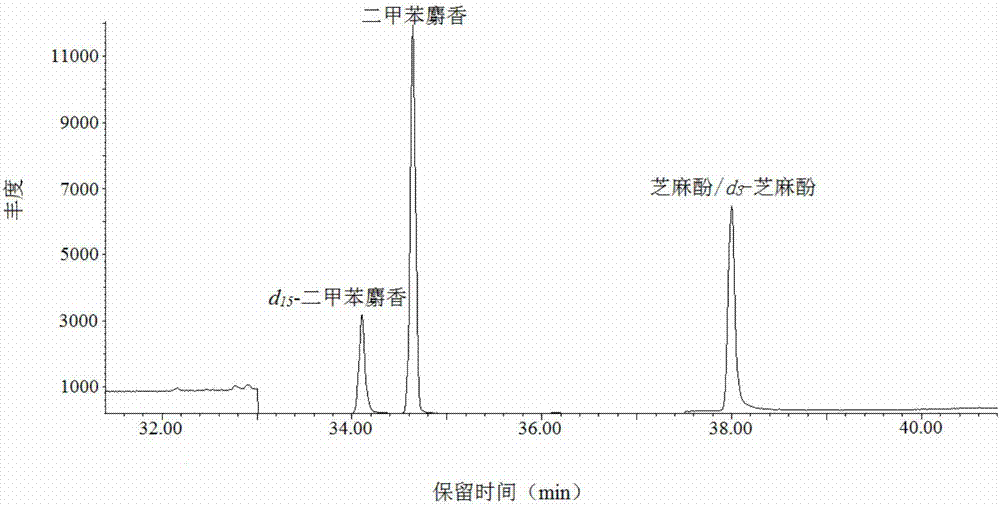
:在卷烟制品的设计和生产过程中往往需要添加各种不同的助剂,如香精和料液、防腐剂、保润剂和助燃剂等,用以改善烟草和卷烟制品理化性能,满足生产、加工需求并提高产品品质。同时,这些添加剂的使用也使之成为影响卷烟产品安全性的重要因素。二甲苯麝香是一种具有麝香气味的人工合成硝基化合物,由于其香味特殊,留香持久的特性,主要被用作化妆品香精及家用洗涤、清洁用品的定香剂。该化合物能够在环境中稳定存在,且具有较大的生物富集性,因此被欧盟列为高度关注物质,国内烟草行业也明确规定烟用添加剂中禁止使用二甲苯麝香。芝麻酚,又名3,4-亚甲二氧基苯酚,存在于芝麻油中,是天然有机酚类化合物,具有较强的抗氧化性。但由于其有一定的致癌性,2006年日本已将芝麻酚列为食品禁用添加剂。上述两种物质目前都已被烟草行业列入禁用添加剂名单,但由于目前我国烟草行业烟用添加剂使用较为广泛、种类较多、来源较复杂,上述有害成分仍有可能在卷烟加香加料过程中进入卷烟产品。因此建立简单、快速、灵敏的测定这些有害物质的分析方法对烟用添加剂的质量控制具有重要意义。目前,这些有害物质的检测方法已有文献报道,例如专利(cn102998382a)采用无水乙醇超声溶解后直接进行气质联用谱仪(gc/ms)检测,但该方法缺少富集手段、灵敏度不高,对于复杂基质中的痕量检测,并不能满足分析要求。分散液液萃取(dispersiveliquid-liquidmicroextraction,dllme)是一种新型的微萃取技术,由rezaee等于2006年首次报道。该方法集采集、萃取、浓缩于一体,相对于一般的液液萃取法而言,具有溶剂用量小、萃取效率高、富集倍数大、操作方便等优点,在痕量分析领域具有广阔的应用前景。然而,常规dllme法也有一定的局限性,该方法一般较为适用于基质为水样的检测,当应用于香精香料、烟用添加剂等乙醇含量较高,有机质较为复杂的样品时,基质效应将不可避免地较为明显,重复性和回收率也有改进的余地。技术实现要素:鉴于以上所述现有技术的缺点,本发明的目的在于提供一种烟用添加剂中二甲苯麝香和芝麻酚的测定方法,对于烟用添加剂中二甲苯麝香和芝麻酚,通过采用在样品中加入同位素内标-分散液液萃取法进行萃取、富集与净化,采用气相色谱质谱联用法进行测定。为实现上述目的及其他相关目的,本发明提供一种烟用添加剂中二甲苯麝香和芝麻酚的测定方法,通过在烟用添加剂中加入待测化合物的同位素内标溶液,进行分散液液萃取,再采用气相色谱-质谱联用法(gc-ms)测定烟用添加剂中二甲苯麝香和芝麻酚的含量。较佳地,所述二甲苯麝香的cas号为81-15-2,所述芝麻酚的cas号为533-31-3。较佳地,所述一种烟用添加剂中二甲苯麝香和芝麻酚的测定方法,具体包括以下步骤:1)标准溶液的配制:配制混合标准储备溶液、内标溶液和混合标准溶液;a1)分别称取二甲苯麝香和芝麻酚的标准品,加入有机溶剂定容,配成混合标准储备溶液;优选地,所述混合标准储备溶液中二甲苯麝香和芝麻酚的浓度均为1-100μg/ml。更优选地,所述混合标准储备溶液中二甲苯麝香和芝麻酚的浓度均为10μg/ml。a2)分别称取二甲苯麝香的同位素和芝麻酚的同位素,加入有机溶剂定容,配成内标溶液;优选地,所述内标溶液中二甲苯麝香的同位素和芝麻酚的同位素的浓度均为10-1000μg/ml。更优选地,所述内标溶液中二甲苯麝香的同位素和芝麻酚的同位素的浓度均为100μg/ml。优选地,所述二甲苯麝香的同位素和芝麻酚的同位素分别为它们所对应的氘代试剂。具体地,所述二甲苯麝香的同位素为氘代二甲苯麝香(d15-二甲苯麝香),所述芝麻酚的同位素为氘代芝麻酚(d3-芝麻酚)。所述氘代二甲苯麝香和氘代芝麻酚为商品化试剂,其化学性质稳定,适于作为内标。a3)分别移取不同体积的步骤a1)中混合标准储备溶液,再分别加入一定体积的步骤a2)中内标溶液,用有机溶剂定容配制为一系列不同浓度的混合标准溶液。优选地,步骤a1)、a2)或a3)中,所述有机溶剂为无水乙醇。优选地,所述混合标准溶液的浓度为0-200ng/ml。更优选地,所述混合标准溶液的浓度为2-200ng/ml。优选地,所述混合标准溶液内,氘代二甲苯麝香和氘代芝麻酚的浓度均为10-1000ng/ml。更优选地,所述混合标准溶液内,氘代二甲苯麝香和氘代芝麻酚的浓度均为100ng/ml。2)样品前处理:称取烟用添加剂样品,加入有机溶剂和步骤1)中的内标溶液,涡旋混匀,进行超声振荡使样品完全溶解后静置,获得提取液;优选地,所述有机溶剂为无水乙醇。优选地,所述烟用添加剂样品加入的质量(克,g)与有机溶剂加入的体积(毫升,ml)之比为1:15-25。更优选地,所述烟用添加剂样品加入的质量(g)与有机溶剂加入的体积(ml)之比为1:20。优选地,所述烟用添加剂样品加入的质量(g)与内标溶液加入的体积(ml)之比为4-6:1。更优选地,所述烟用添加剂样品加入的质量(g)与内标溶液加入的体积(ml)之比为5:1。优选地,所述烟用添加剂样品中还加入水。所述水为去离子水。当烟用添加剂样品为粘稠样品时,加入水有助于分散样品。优选地,所述烟用添加剂样品加入的质量(g)与水加入的体积(ml)之比为1:0-4。更优选地,所述烟用添加剂样品加入的质量(g)与水加入的体积(ml)之比为1:0-2。优选地,所述涡旋时间为1±0.1min。所述涡旋作为一种样品前处理方式,主要用于将溶质与萃取溶剂之间进行充分混合,提升萃取效率。优选地,所述超声振荡时间为9-11min。更优选地,所述超声振荡时间为10min。优选地,所述静置时间为9-11min。更优选地,所述静置时间为10min。3)分散液液萃取:取步骤2)中提取液,加入水、萃取剂和分散剂,充分混匀,进行超声萃取从而形成微乳液,再进行离心后,取下层沉积相,待测;优选地,所述提取液与水之间加入的体积比为1:5-7。更优选地,所述提取液与水之间加入的体积比为1:6。所述水为去离子水。优选地,所述萃取剂选自四氯化碳、氯仿和二氯甲烷中的一种。更优选地,所述萃取剂为二氯甲烷。优选地,所述提取液与萃取剂之间加入的体积比为5:1-2。更优选地,所述提取液与萃取剂之间加入的体积比为5:1。优选地,所述分散剂选自甲醇、丙酮和乙腈中的一种。更优选地,所述分散剂为丙酮。优选地,所述提取液与分散剂之间加入的体积比为:2:1-3。更优选地,所述提取液与分散剂之间加入的体积比为1:1。优选地,所述超声萃取时间为9-11min。更优选地,所述超声萃取时间为10min。优选地,所述离心条件为:离心时间为:4-6min;离心转速:3000-5000rpm。更优选地,所述离心条件为:离心时间为:5min;离心转速:4000rpm。优选地,所述下层沉积相通过微量进样器提取,转移至装有内插管的色谱进样瓶,待测。步骤3)中分散液液萃取的具体过程见图1。4)样品定性检测:分别将步骤1)配制的混合标准溶液和步骤3)中待测的下层沉积相进行gc-ms检测,比较保留时间,通过谱图数据库检索,进行定性,确定二甲苯麝香和芝麻酚成分;所述比较保留时间是指,通过标准溶液中各组分和内标的保留时间,与实际样品中各组分和内标的保留时间,对比分析是否一致。所述谱图数据库检索是指,将全扫描(scan)模式下的全扫描特征离子碎片谱图与质谱标准谱图数据库检索结果进行比较,对比分析是否一致。优选地,所述谱图数据库检索条件为:质量扫描范围:35-300amu;谱图数据库:wiley标准谱库;成分判断依据选择匹配度≥80。5)样品定量检测:分别将步骤1)配制的混合标准溶液和步骤3)中待测的下层沉积相进行gc-ms检测,采用内标标准曲线法进行定量,获得下层沉积相中二甲苯麝香和芝麻酚的含量;优选地,所述内标标准曲线法包括以下步骤:b1)将步骤1)的a3)中一系列不同浓度的混合标准溶液分别进行gc-ms检测,获得目标物/内标物的色谱峰面积比与相应目标物/内标物的浓度比的线性关系,绘制相应的标准工作曲线,计算得到各标准工作曲线的回归方程;所述目标物为二甲苯麝香和芝麻酚的标准品,所述内标物为二甲苯麝香和芝麻酚的同位素。更优选地,所述标准曲线中,以目标物/内标物的色谱峰面积比为纵坐标(y轴),其目标物/内标物的浓度比为横坐标(x轴)。b2)将步骤3)中待测的下层沉积相进行gc-ms检测,将获得的二甲苯麝香和芝麻酚与相应内标物的色谱峰面积比,代入步骤b1)中各标准工作曲线的回归方程,并根据内标物的已知浓度,计算得到下层沉积相中二甲苯麝香和芝麻酚的浓度。更优选地,所述目标物/内标物的浓度为质量浓度。进一步的,所述目标物/内标物的浓度比为质量浓度比。优选地,所述步骤4)或5)中gc-ms检测条件为:气相色谱条件为:色谱柱:hp-innowax熔融石英毛细柱(30m×0.25mm,0.25μm);进样口温度:250℃;进样量:1μl;载气:高纯氦气,载气纯度≥99.999%;载气流速:1.0ml/min,恒流模式;分流比:10:1;升温程序:初始温度100℃保持1min,以3℃/min的速率升至240℃,保持5min,即质谱条件为:传输线温度:260℃;离子源温度230℃;四级杆温度150℃;溶剂延迟:10min;电离方式:电子轰击源(ei源);电离能量:70ev;扫描方式:全扫描(scan)或选择离子监测(sim)。6)实际样品含量的测定:将步骤5)获得的下层沉积相中二甲苯麝香和芝麻酚的含量分别代入公式,计算得到烟用添加剂样品中二甲苯麝香和芝麻酚的含量。优选地,所述公式为c0=(csed×v0)/vsed,其中,c0为烟用添加剂样品中二甲苯麝香或芝麻酚的含量,μg/l;csed为下层沉积相中二甲苯麝香或芝麻酚的含量,μg/l;v0为烟用添加剂样品的体积,ml;vsed为下层沉积相的体积,ml。如上所述,本发明首次开发了一种烟用添加剂中二甲苯麝香和芝麻酚的测定方法,采用将分散液液萃取技术结合同位素内标法,对烟用添加剂中的二甲苯麝香和芝麻酚进行萃取、富集,并采用气相色谱质谱联用法对其含量进行测定。该方法采用的分散液液萃取技术,集采样、萃取和浓缩于一体,具有溶剂用量小,萃取效率高,富集倍数大的优点。此外,由于采用了同位素内标,可更好地补偿液液萃取过程中的样品损失并减少质谱离子化过程中的基质效应,使得本方法相比较常规的分散液相微萃取(dllme)法的检测结果更为准确、可靠,适用范围更广。该方法采用标准加入法定性、内标标准曲线法定量,二甲苯麝香和芝麻酚的标准曲线的线性关系良好,相关系数r2分别为0.9986、0.9995,方法检出限(lod)分别为0.39、0.67μg/l,定量检出限(loq)分别为1.18、2.11μg/l,具有较高的灵敏度。方法的回收率为90.0%-100.4%之间,平均相对标准偏差(rsd%)在2.0%~7.2%之间,具有较好的准确性和精密度。本方法操作简便、绿色环保、灵敏度高,能够实现了对烟用添加剂中痕量二甲苯麝香和芝麻酚的准确有效测定,可用于烟用添加剂的质量控制。附图说明图1显示为本发明的分散液液微萃取流程图。图2显示为本发明的标准溶液选择离子色谱图。图3显示为本发明的不同萃取溶剂对萃取效率的影响示意图。图4显示为本发明的不同分散剂对萃取效率的影响示意图。图5显示为本发明的萃取剂体积对萃取效率的影响示意图。图6显示为本发明的分散剂体积对萃取效率的影响示意图。图7显示为本发明的实施例中3#样品溶液选择离子色谱图。具体实施方式下面结合具体实施例进一步阐述本发明,应理解,这些实施例仅用于说明本发明而不用于限制本发明的保护范围。以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。以下实施例中使用的试剂及仪器均为常规使用的试剂及仪器,均可从市场上购买获得,具体如下:1、试剂烟用添加剂:浸膏、香精、料液(上海牡丹香精香料公司);二甲苯麝香(纯度为99.7%,甲醇为溶剂,上海安谱);芝麻酚(纯度为98%,百灵威科技);氘代二甲苯麝香(100ng/μlinacetone,dr.ehrenstorfergmbh,germany);氘代芝麻酚(>98.3%,cdn,canada);去离子水(纯水机自制);无水乙醇、氯仿、二氯甲烷、甲醇、丙酮、乙腈(均为hplc级,纯度>99.9%,从merckchemicals购入);四氯化碳(分析纯,>99.5%,上海经纬化工)。2、仪器气相色谱-质谱联用仪(7890b/5977c,agilenttechnologies,usa);涡旋混合仪(talboysstdvortexmixer,henrytroemner,llc,usa);超声振荡仪(sw30h,sonoswiss,switzerland);离心机(rj-ldl-50g,无锡瑞江);微量进样器(100μl/250μl,agilenttechnologies,usa)。实施例11、标准溶液的配制分别称取二甲苯麝香和芝麻酚的标准品,加入无水乙醇定容,配成混合标准储备溶液。混合标准储备溶液中二甲苯麝香和芝麻酚的浓度均为1-100μg/ml。同时,分别移取一定体积的二甲苯麝香的同位素和芝麻酚的同位素,具体为氘代二甲苯麝香和氘代芝麻酚,加入无水乙醇定容,配成内标溶液,内标溶液中二甲苯麝香的同位素和芝麻酚的同位素的浓度为10-1000μg/ml。分别准确移取不同体积混合标准储备溶液,置于多个100ml容量瓶中,再分别准确加入一定体积的内标溶液,用无水乙醇定容,配制为一系列混合标准溶液。配制的系列混合标准溶液浓度为0-200ng/ml,其中内标物浓度为10-1000ng/ml。2、样品前处理称取烟用添加剂样品,加入有机溶剂和内标溶液,涡旋0.9-1.1min混匀,进行超声振荡9-11min使样品完全溶解后静置9-11min,获得提取液。其中,有机溶剂为无水乙醇,烟用添加剂样品加入的质量(g)与有机溶剂加入的体积(ml)之比为1:15-25,烟用添加剂样品加入的质量(g)与内标溶液加入的体积(ml)之比为4-6:1。当烟用添加剂样品为粘稠样品时,需加入水以助分散,水为去离子水,烟用添加剂样品加入的质量(g)与水加入的体积(ml)之比为1:0-4。取一定体积的提取液,加入水、萃取剂和分散剂,充分混匀,进行超声萃取9-11min从而形成微乳液,再在离心转速为3000-5000rpm条件下进行离心4-6min后,通过微量进样器提取下层沉积相,转移至装有内插管的色谱进样瓶,待测,具体过程见图1。其中,提取液与水之间加入的体积比为1:5-7。萃取剂选自四氯化碳、氯仿和二氯甲烷中的一种,提取液与萃取剂之间加入的体积比为5:1-2。分散剂选自甲醇、丙酮和乙腈中的一种,提取液与分散剂之间加入的体积比为:2:1-3。3、样品含量的测定分别将上述步骤1配制的标准溶液和步骤2中待测的下层沉积相进行gc-ms检测,比较保留时间,通过谱图数据库检索,进行定性,确定二甲苯麝香和芝麻酚成分。其中,质量扫描范围为30-350amu,谱图数据库为wiley标准谱库;成分判断依据选择匹配度≥80。同时采用内标标准曲线法进行定量,获得下层沉积相中二甲苯麝香和芝麻酚的浓度含量。再通过下层沉积相中二甲苯麝香和芝麻酚的浓度含量,计算得到烟用添加剂样品中二甲苯麝香和芝麻酚的含量。具体来说,内标标准曲线法是先将上述步骤1中一系列不同浓度的标准溶液分别进行gc-ms检测,获得二甲苯麝香和芝麻酚成分/内标物的色谱峰面积比与相应二甲苯麝香和芝麻酚成分/内标物的浓度比的线性关系,绘制相应的标准工作曲线,以二甲苯麝香和芝麻酚成分与内标物的色谱峰面积比为纵坐标(y轴),其相应二甲苯麝香和芝麻酚成分与内标物的浓度比为横坐标(x轴),计算得到各标准工作曲线的回归方程。再将上述步骤2中待测的下层沉积相进行gc-ms检测,将获得的二甲苯麝香和芝麻酚成分与内标物的峰面积比,代入各标准工作曲线的回归方程,并根据内标物的已知浓度,计算得到待测的下层沉积相中二甲苯麝香和芝麻酚的浓度。最后将获得的下层沉积相中二甲苯麝香和芝麻酚的浓度分别代入公式c0=(csed×v0)/vsed,其中,c0为烟用添加剂样品中二甲苯麝香或芝麻酚的含量,μg/l;csed为下层沉积相中二甲苯麝香或芝麻酚的含量,μg/l;v0为烟用添加剂样品的体积,ml;vsed为下层沉积相的体积,ml;从而计算得到烟用添加剂样品中二甲苯麝香和芝麻酚的含量。这里所述二甲苯麝香、芝麻酚和内标物的浓度为质量浓度。其中,gc-ms检测条件为:气相色谱条件为:色谱柱:hp-innowax熔融石英毛细柱(30m×0.25mm,0.25μm);进样口温度:250℃;进样量:1μl;载气:高纯氦气,载气纯度≥99.999%;载气流速:1.0ml/min,恒流模式;分流比:10:1;升温程序:初始温度100℃保持1min,以3℃/min的速率升至240℃,保持5min,即质谱条件为:传输线温度:260℃;离子源温度230℃;四级杆温度150℃;溶剂延迟:10min;电离方式:电子轰击源(ei源);电离能量:70ev;扫描方式:全扫描(scan)或选择离子监测(sim)。实施例21、标准溶液的配制分别称取二甲苯麝香和芝麻酚的标准品,加入无水乙醇定容,配成混合标准储备溶液。混合标准储备溶液中二甲苯麝香和芝麻酚的浓度为10μg/ml。同时,分别移取一定体积的二甲苯麝香的同位素和芝麻酚的同位素,具体为氘代二甲苯麝香和氘代芝麻酚,加入无水乙醇定容,配成内标溶液,内标溶液中二甲苯麝香的同位素和芝麻酚的同位素的浓度为100μg/ml。分别准确移取不同体积混合标准储备溶液,置于5个100ml容量瓶中,再分别准确加入一定体积的内标溶液,用无水乙醇定容,配制为一系列混合标准溶液。配制的系列混合标准溶液浓度分别为2ng/ml、20ng/ml、50ng/ml、100ng/ml、200ng/ml,其中内标物浓度为100ng/ml。2、样品前处理称取0.5g烟用添加剂样品,加入10ml无水乙醇和0.1ml内标溶液,涡旋1min混匀,进行超声振荡10min使样品完全溶解后静置10min,获得提取液。当烟用添加剂样品为粘稠样品时,需加入1ml去离子水以助分散。取0.5ml的提取液20ml于尖底玻璃离心试管,加入3ml去离子水、100μl二氯甲烷和0.5ml丙酮,充分混匀,进行超声萃取10min从而形成微乳液,再在离心转速为4000rpm条件下进行离心5min后,含待测化合物的沉积相沉淀在试管底部,通过微量进样器提取下层沉积相,记录其体积,转移至装有内插管的色谱进样瓶,待测,具体过程见图1。3、样品含量的测定分别将上述步骤1配制的标准溶液和步骤2中待测的下层沉积相进行gc-ms检测,比较保留时间,通过谱图数据库检索,进行定性,确定二甲苯麝香和芝麻酚成分。其中,质量扫描范围为30-350amu,谱图数据库为wiley标准谱库;成分判断依据选择匹配度≥80。同时采用内标标准曲线法进行定量,获得下层沉积相中二甲苯麝香和芝麻酚的浓度含量。再通过下层沉积相中二甲苯麝香和芝麻酚的浓度含量,计算得到烟用添加剂样品中二甲苯麝香和芝麻酚的含量。具体来说,内标标准曲线法是先将上述步骤1中一系列不同浓度的标准溶液分别进行gc-ms检测,获得二甲苯麝香和芝麻酚成分/内标物的色谱峰面积比与相应二甲苯麝香和芝麻酚成分/内标物的浓度比的线性关系,绘制相应的标准工作曲线,以二甲苯麝香和芝麻酚成分与内标物的色谱峰面积比为纵坐标(y轴),其相应二甲苯麝香和芝麻酚成分与内标物的浓度比为横坐标(x轴),计算得到各标准工作曲线的回归方程。再将上述步骤2中待测的下层沉积相进行gc-ms检测,将获得的二甲苯麝香和芝麻酚成分与内标物的峰面积比,代入各标准工作曲线的回归方程,并根据内标物的已知浓度,计算得到待测的下层沉积相中二甲苯麝香和芝麻酚的浓度。最后将获得的下层沉积相中二甲苯麝香和芝麻酚的浓度分别代入公式c0=(csed×v0)/vsed,其中,c0为烟用添加剂样品中二甲苯麝香或芝麻酚的含量,μg/l;csed为下层沉积相中二甲苯麝香或芝麻酚的含量,μg/l;v0为烟用添加剂样品的体积,ml;vsed为下层沉积相的体积,ml;从而计算得到烟用添加剂样品中二甲苯麝香和芝麻酚的含量。这里所述二甲苯麝香、芝麻酚和内标物的浓度为质量浓度。二甲苯麝香、芝麻酚和2种相应同位素内标物的保留时间和特征离子见表1,图2。表1二甲苯麝香、芝麻酚及其同位素内标物的保留时间和定性定量离子序号化合物名称保留时间(min)定量离子(m/z)定性离子(m/z)1二甲苯麝香34.882822972氘代二甲苯麝香34.082943123芝麻酚37.97138524氘代芝麻酚37.9714054其中,gc-ms检测条件为:气相色谱条件为:色谱柱:hp-innowax熔融石英毛细柱(30m×0.25mm,0.25μm);进样口温度:250℃;进样量:1μl;载气:高纯氦气,载气纯度≥99.999%;载气流速:1.0ml/min,恒流模式;分流比:10:1;升温程序:初始温度100℃保持1min,以3℃/min的速率升至240℃,保持5min,即质谱条件为:传输线温度:260℃;离子源温度230℃;四级杆温度150℃;溶剂延迟:10min;电离方式:电子轰击源(ei源);电离能量:70ev;扫描方式:全扫描(scan)或选择离子监测(sim)。实施例3根据实施例2中测定烟用添加剂样品中二甲苯麝香和芝麻酚的含量的方法步骤,分别选用二氯甲烷、氯仿和四氯化碳三种溶剂作为萃取剂进行样品前处理,测定结果见图3。由图3可知,二氯甲烷、氯仿和四氯化碳均可作为萃取剂进行样品前处理,但其中二氯甲烷的萃取效果最佳。实施例4根据实施例2中测定烟用添加剂样品中二甲苯麝香和芝麻酚的含量的方法步骤,分别选用甲醇、丙酮和乙腈三种溶剂作为分散剂进行样品前处理,测定结果见图4。由图4可知,甲醇、丙酮和乙腈均可作为分散剂进行样品前处理,但其中丙酮的分散效果最佳。实施例5根据实施例2中测定烟用添加剂样品中二甲苯麝香和芝麻酚的含量的方法步骤,分别选用50、100、150、200μl的二氯甲烷作为萃取剂进行样品前处理,测定结果见图5。由图5可知,当选用100μl的二氯甲烷作为萃取剂进行样品前处理时,萃取效果最佳。实施例6根据实施例2中测定烟用添加剂样品中二甲苯麝香和芝麻酚的含量的方法步骤,分别选用0、0.25、0.5、0.75、1ml的丙酮作为分散剂进行样品前处理,测定结果见图6。由图6可知,当选用0.5ml的丙酮作为分散剂进行样品前处理时,分散效果最佳。实施例7分别称取二甲苯麝香和芝麻酚的标准品,加入无水乙醇定容,配成混合标准储备溶液。混合标准储备溶液中二甲苯麝香和芝麻酚的浓度为10μg/ml同时,分别移取一定体积的二甲苯麝香的同位素和芝麻酚的同位素,具体为氘代二甲苯麝香和氘代芝麻酚,加入无水乙醇定容,配成内标溶液,内标溶液中二甲苯麝香的同位素和芝麻酚的同位素的浓度为100μg/ml。分别准确移取不同体积混合标准储备溶液,置于多个100ml容量瓶中,再分别准确加入一定体积的内标溶液,用无水乙醇定容,配制为一系列混合标准溶液。配制的系列混合标准溶液浓度分别为2ng/ml、20ng/ml、50ng/ml、100ng/ml、200ng/ml,其中内标物浓度为100ng/ml。将上述系列混合标准溶液按本文图1所述步骤进行dllme(分散液液萃取)处理,然后gc/ms上机分析,采用内标法定量,以二甲苯麝香和芝麻酚成分与内标物的色谱峰面积比为纵坐标(y轴),其相应二甲苯麝香和芝麻酚成分与内标物的浓度比为横坐标(x轴),进行回归分析,得到二甲苯麝香和芝麻酚的回归方程及其相关系数,如表2所示。表2待测化物线性回归方程、回归系数、检测限以及富集倍数注:y为峰面积比;x为浓度比由表2可知,二甲苯麝香和芝麻酚标准曲线的线性关系良好,二甲苯麝香的相关系数r2=0.9986,芝麻酚的相关系数r2=0.9995。将最低浓度的标准溶液直接进行gc/ms分析,以3倍信噪比(s/n=3)为方法的检出限(mdl或lod),10倍信噪比(s/n=10)为定量限(mql或loq),则二甲苯麝香的lod和loq分别为0.39和1.18μg/l,而芝麻酚的lod和loq分别为0.67和2.11μg/l,具有较高的灵敏度。同时,根据以下公式ⅰ、ⅱ计算富集倍数(ef)和萃取回收率(er),其中,公式ⅰ为:公式ⅱ为:式中,c0为烟用添加剂样品中二甲苯麝香或芝麻酚的含量,μg/l;csed为下层沉积相中二甲苯麝香或芝麻酚的含量,μg/l;v0为烟用添加剂样品的体积,ml;vsed为下层沉积相的体积,ml;结果详见表2。由公式ⅰ、ⅱ和表2可知,二甲苯麝香和芝麻酚的富集倍数(富集因子)分别为265和298,富集倍数非常大,由于富集倍数(富集因子)与方法灵敏度呈正比,该处理方法灵敏度高。同时,由于萃取回收率是评价分析方法的可行性与否的指标,一般在80-120%之间,而二甲苯麝香和芝麻酚的萃取回收率分别为106.2%和119.1%,落在范围内,该分析方法的可行性高。实施例8选取1#(浸膏,粘稠状),2#(香精)和3#(料液)三种代表性烟用添加剂样品,分别称取0.5g于离心管中,其中1#和3#样品加1ml水,再加入10ml无水乙醇和0.1ml内标溶液,涡旋1min混匀,进行超声振荡10min使样品完全溶解后静置10min,获得提取液。取0.5ml的提取液于20ml尖底玻璃离心试管,加入3ml去离子水,将100μl二氯甲烷和0.5ml丙酮使用微量进样器快速注入,充分混匀,进行超声萃取10min从而形成微乳液,再在离心转速为4000rpm条件下进行离心5min后,含待测化合物的沉积相沉淀在试管底部,通过微量进样器提取下层沉积相,记录其体积,转移至装有内插管的色谱进样瓶,待测,具体过程见图1。实施例9将实施例8中制备的1#和2#样品分别进行三个水平的加标回收率实验,进行了精密度测定,每个添加水平平行测定5次,回收率和相对标准偏差(rsd%)见表3。由表3可知,二甲苯麝香和芝麻酚的回收率在90.0%~100.4%之间,rsd%在2.0%~7.2%之间,准确性和重复性较好,可以满足定量需要。表3实际样品中待测化合物加标回收率和相对标准偏差实施例10为了进一步验证本方法的准确性,分别选用本实施例8中1#、2#、3#样品,采用本发明中方法与专利cn102998382a中测定方法对其进行了测试,将两种方法所得测定结果进行对比,详见表4。其中,本发明中方法测定3#样品的色谱图见图7。由表4可知,专利方法未能检出的1#、2#、3#样品中的二甲苯麝香和3#样品中的芝麻酚,而本发明中的方法均能够检测到。2#样品中的芝麻酚,用两种方法均未能测出,并且1#样品中的芝麻酚,用两种方法均能测出,且检测结果非常接近,证明本方法的准确性能够满足要求,同时富集效果明显。表4两种方法测定结果比较备注*:“—”表示未检出所以,本发明有效克服了现有技术中的种种缺点而具高度产业利用价值。上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属
技术领域:
中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1