润滑油组合物的利记博彩app
本发明涉及温度粘度特性和低温粘度特性优异、并且具有极其优异的剪切稳定性的润滑油组合物。
背景技术:
对于齿轮油、变速箱油(transmission oils)、液压油、润滑脂(greases)这样的润滑油,不仅要求对内燃机和机床的保护、散热这样的性能,而且还要求耐磨损性、耐热性、耐淤渣性(sludge resistance)、润滑油消耗特性、节省燃耗性等多种性能。而且,近年来,伴随着所使用的内燃机、工业机械的高性能化、高输出功率化、运转条件的严酷化等,各要求性能日益增高。尤其是近来,润滑油的使用环境愈发严酷,但另一方面,出于对环境问题的考虑,有要求延长润滑油寿命的倾向,从而不仅要求提高耐热性、提高氧化稳定性,而且还要求抑制由来自发动机、机械的剪切应力导致的低粘度化、即提高润滑油的剪切稳定性。另外,另一方面,为了提高发动机的能量转换效率、或确保极低温环境下的发动机的良好的润滑性,在高温下保持润滑油的油膜、在低温下进一步保持流动性这样的温度粘度特性被认为是重要的。对于此处所述的温度粘度特性而言,作为一个指标,可通过利用JIS K2283中记载的方法算出的粘度指数进行量化,显示更高的粘度指数的润滑油具有更优异的温度粘度特性。
因此,对于润滑油而言,要求耐热性、氧化稳定性及剪切稳定性优异、且具有良好的温度粘度特性的材料。
尤其是,对于被用于汽车的润滑油、即差动齿轮油之类的汽车用齿轮油以及以变速箱油为代表的驱动油等,逐渐要求现有水平以上的优异的温度粘度特性、以及-40℃这样的极低温度下的高流动性、即优异的低温粘度特性。上述粘度特性直接关系到汽车的燃油效率性能,之所以要求提高该性能,是因为在1997年通过京都议定书以后,近年来世界各国政府确立了针对载客车的二氧化碳排放规定、燃油效率规定、或未来目标。
基于此,为了提高燃油效率以达成燃油效率目标,载客车发动机的各部分日趋小型化,所使用的润滑油量也逐渐减少。因此,润滑油所承受的负荷逐渐增大,日益要求进一步延长润滑油寿命。
汽车用齿轮油或变速箱油承受来自齿轮或金属带等的剪切应力,因此,在使用过程中,润滑油中所使用的基材的分子被切断,由此导致润滑油粘度降低。若润滑油粘度降低,则发生齿轮彼此、金属间的接触,对齿轮造成明显的损伤。因此,需要预先预测使用期间的粘度降低,并预先提高制造润滑油时的粘度(初始粘度),由此设计为伴随使用而发生劣化后的润滑油能够进行理想的润滑。在汽车工程师协会(Society of Automotive Engineers,SAE)制定的汽车齿轮油的粘度标准J306中,确定了CRC L-45-T-93中规定的剪切试验(方法C(method C),20小时)后的最低粘度。
当然,若润滑油中使用的基材的剪切稳定性优异,则可延长润滑油的寿命,无需提高润滑油的初始粘度,结果,可降低润滑油相对于齿轮的搅拌阻力,因此,可实现燃油效率的提高。
另外,若温度粘度特性、即润滑油粘度的温度依赖性低,则在低温环境下也可抑制润滑油的粘度上升,结果,与现有技术相比,由润滑油导致的齿轮阻力相对地降低,可实现燃油效率的提高。
此外,作为近年来的提高燃油效率的对策,通过将差动齿轮油或变速箱油的粘度降低至以往水平以下而实现了降低由润滑油导致的搅拌阻力,结果齿轮中的金属接触的危险性愈发增高,因此,要求不引起粘度降低的、剪切稳定性极高的材料。
基于上述性能提高的要求,针对通常以20小时的试验时间进行的CRC L-45-T-93剪切试验,J306中还同样规定了每种驱动油在以相当于通常试验时间的5倍的试验时间即100小时进行试验后的最低粘度,并开始要求维持该最低粘度。
作为满足上述这样的要求的润滑油基础油,在工业上广泛使用作为合成润滑油的聚-α-烯烃(PAO)。如专利文献1~3等中记载那样,这样的PAO可通过利用酸催化剂使高级α-烯烃进行低聚反应而得到。
另一方面,如专利文献4中记载那样,已知乙烯-α-烯烃共聚物也与PAO同样,可用作粘度指数、氧化稳定性、剪切稳定性、耐热性优异的合成润滑油。
作为可用作合成润滑油的乙烯-α-烯烃共聚物的制造方法,以往,一直采用专利文献5及专利文献6中记载那样的利用包含钒化合物和有机铝化合物的钒系催化剂的方法。作为这样的乙烯-α-烯烃共聚物,尤其是主要使用乙烯-丙烯共聚物。
另外,作为以高聚合活性制造共聚物的方法,已知专利文献7、专利文献8中记载那样的使用包含二茂锆(zirconocene)等茂金属化合物和有机铝氧化合物(铝氧烷)的催化剂体系的方法等,专利文献9中公开了一种合成润滑油的制造方法,所述合成润滑油包含乙烯-α-烯烃共聚物,所述乙烯-α-烯烃共聚物是通过使用组合特定的茂金属催化剂和铝氧烷而成的催化剂体系而得到的。
近年来,对作为低温粘度特性及耐热性·氧化稳定性优异的合成润滑油基材的PAO或乙烯-丙烯共聚物等的需求存在增大的倾向,从节省燃耗·节约能源的观点考虑,要求进一步改善粘度指数、低温粘度特性。
基于上述要求,发明了通过专利文献10~13中记载那样的使用包含二茂锆等茂金属化合物和有机铝氧化合物(铝氧烷)的催化剂体系的方法等而得到的PAO。
然而,以往,润滑油组合物的剪切稳定性依赖于含有的成分的分子量是已知的。也就是说,含有分子量较高的成分的润滑油组合物容易发生由剪切应力导致的粘度降低,该粘度降低率与含有成分的分子量相关。
另一方面,通过含有分子量较高的成分,可提高润滑油组合物的温度粘度特性、低温粘度特性。即,对于用于润滑油组合物的PAO或乙烯-丙烯共聚物等而言,存在下述二律背反(trade-off)关系:随着分子量增高,温度粘度特性提高,但剪切稳定性降低。关于该方面,从同时实现剪切稳定性和温度粘度特性这样的观点考虑,存在改良的余地。
现有技术文献
专利文献
专利文献1:美国专利第3,780,128号说明书
专利文献2:美国专利第4,032,591号说明书
专利文献3:日本特开平1-163136号公报
专利文献4:日本特开昭57-117595号公报
专利文献5:日本特公平2-1163号公报
专利文献6:日本特公平2-7998号公报
专利文献7:日本特开昭61-221207号公报
专利文献8:日本特公平7-121969号公报
专利文献9:日本专利第2796376号公报
专利文献10:日本特开2001-335607号公报
专利文献11:日本特表2004-506758号公报
专利文献12:日本特表2009-503147号公报
专利文献13:日本特表2009-514991号公报
技术实现要素:
发明所要解决的课题
鉴于这样的现有技术中的问题点,从汽车、工业用机械的节省燃耗及节约能源的观点考虑,本发明所要解决的课题在于提供兼具极其优异的剪切稳定性和低温粘度特性的润滑油。
用于解决课题的手段
本申请的发明人们为了开发具有优异性能的润滑油组合物而进行了深入研究,结果发现,含有特定的润滑油基础油和特定的α-烯烃(共)聚合物、且满足特定条件的润滑油组合物可解决上述课题,从而完成了本发明。
本申请的发明人们按照CRC L-45-T-93中记载的方法,通过将试验时间设为100小时的剪切试验,发现了润滑油组合物的特定的分子量区域受到影响。基于此对润滑油组合物进行优化,从而发明了兼具高剪切稳定性、温度粘度特性和低温粘度特性的润滑油组合物。具体而言,可举出以下的方案。
〔1〕润滑油组合物,其含有:
润滑油基础油(A),所述润滑油基础油(A)在100℃时的运动粘度为1~10mm2/s;和
乙烯-α-烯烃共聚物(B),所述乙烯-α-烯烃共聚物(B)具有以下的(B1)~(B4)的特征,
所述润滑油组合物在100℃时的运动粘度为20mm2/s以下,
所述润滑油组合物在3,000~10,000的分子量范围内具有峰顶(peak top),所述分子量是利用凝胶渗透色谱法(GPC)、按照标准聚苯乙烯换算而得到的,
在具有形成该峰顶的分子量以上的高分子量的成分中,按照标准聚苯乙烯换算得到的分子量为20,000以上的成分的重量分数为1~10%,
(B1)利用凝胶渗透色谱法(GPC)测得的分子量中,按照标准聚苯乙烯换算得到的峰顶分子量为3,000~10,000,
(B2)不具有利用差热量热计(DSC)测得的熔融峰,
(B3)下述式[1]表示的B值为1.1以上,
[数学式1]
(式中,PE表示乙烯成分所占的摩尔分数,PO表示α-烯烃成分所占的摩尔分数,POE表示全部二单元组链(dyad sequences)的乙烯-α-烯烃链的摩尔分数。)
(B4)100℃时的运动粘度为140~500mm2/s。
〔2〕如项〔1〕所述的润滑油组合物,其中,上述乙烯-α-烯烃共聚物(B)的乙烯摩尔含有率在30~70mol%的范围内。
〔3〕如项〔1〕或项〔2〕所述的润滑油组合物,其中,上述乙烯-α-烯烃共聚物(B)的α-烯烃为丙烯。
〔4〕如项〔1〕~〔3〕中任一项所述的润滑油组合物,其是汽车用润滑油组合物。
〔5〕汽车用变速箱油,其包含项〔4〕所述的润滑油组合物,所述润滑油组合物的100℃运动粘度为7.5mm2/s以下。
发明的效果
本发明的润滑油组合物是与以往的润滑油相比兼具极其优异的剪切稳定性和高温度粘度特性、以及优异的低温粘度特性的润滑油组合物,可合适地应用于汽车用润滑油、汽车用变速箱油、尤其是汽车用齿轮油、及汽车用低粘度变速箱油。
附图说明
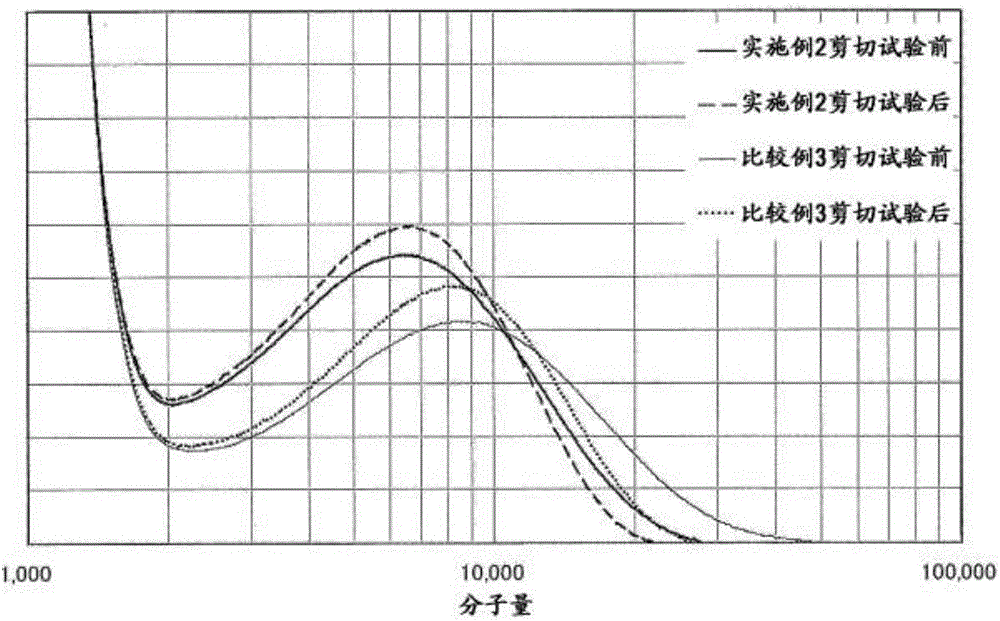
[图1]为实施例2和比较例3中的润滑油组合物的剪切试验前(实线)与剪切试验后(虚线)的GPC图谱的比较。
[图2]为图1中的GPC图谱的分子量10,000附近的放大图。
具体实施方式
以下,对本发明涉及的润滑油组合物进行详细说明。
<(A)润滑油基础油>
作为润滑油基础油(A),除了100℃时的运动粘度为1~10mm2/s之外,没有特别限制,可使用用于通常的润滑油的矿物油系润滑油基础油及/或合成系润滑油基础油(以下,也称为“合成烃油”。)。
作为矿物油系润滑油基础油,根据纯化的手段不同存在几个等级,具体而言,可例举:针对将常压残余油(所述常压残余油通过对原油进行常压蒸馏而得到)减压蒸馏而得到的润滑油馏分,进行溶剂脱沥青、溶剂萃取、氢化分解、溶剂脱蜡、氢化纯化等处理中的1种以上从而进行纯化而得到的产物;或蜡异构化矿物油等润滑油基础油。
另外,利用费-托法得到的天然气制(Gas-to-liquid,GTL)基础油也是可合适地使用的润滑油基础油。这样的GTL基础油例如被记载于EP0776959、EP0668342、WO97/21788、WO00/15736、WO00/14188、WO00/14187、WO00/14183、WO00/14179、WO00/08115、WO99/41332、EP1029029、WO01/18156及WO01/57166中。
作为合成烃油,例如,可举出α-烯烃低聚物、烷基苯类、烷基萘类、异丁烯低聚物或其氢化物、链烷烃类(paraffins)、聚氧亚烷基二醇、二烷基二苯基醚、聚苯醚、脂肪酸酯等。
其中,作为α-烯烃低聚物,可使用选自碳原子数为8~12的烯烃中的至少1种烯烃的低分子量低聚物(不包括乙烯-α-烯烃共聚物(B)。)。若本发明的润滑油组合物中使用α-烯烃低聚物,则可得到温度粘度特性、低温粘度特性、以及耐热性极其优异的润滑油组合物。这样的α-烯烃低聚物可通过以齐格勒催化剂、路易斯酸为催化剂的阳离子聚合、热聚合、自由基聚合进行制造。在工业上也可获得,在市场上有100℃运动粘度为2mm2/s~100mm2/s的α-烯烃低聚物出售。例如,可举出NESTE公司制NEXBASE、ExxonMobil Chemical公司制Spectrasyn、Ineos Oligmers公司制Durasyn、Chevron Phillips Chemical公司制Synfluid等。
烷基苯类、烷基萘类通常大部分是烷基链长为6~14个碳原子的二烷基苯或二烷基萘,这样的烷基苯类或烷基萘类可通过苯或萘与烯烃的傅-克烷基化反应进行制造。烷基苯类或烷基萘类的制造中使用的烷基化烯烃可以是线状或分支状的烯烃或者它们的组合。它们的制造方法例如被记载于美国专利第3,909,432号说明书中。
作为脂肪酸酯,没有特别限制,可举出以下这样的仅由碳、氧、氢形成的脂肪酸酯。
可举出由一元酸和醇制造的单酯;由二元酸和醇、或由二元醇和一元酸或酸混合物制造的二酯;使二元醇、三元醇(例如三羟甲基丙烷)、四元醇(例如季戊四醇)、六元醇(例如二季戊四醇)等与一元酸或酸混合物反应而制造的多元醇酯等。作为这些酯的例子,可举出戊二酸二(十三烷基酯)、己二酸二-2-乙基己酯、己二酸二异癸酯、己二酸二(十三烷基酯)、癸二酸二-2-乙基己酯、壬酸十三烷基酯、己二酸二-2-乙基己酯、壬二酸二-2-乙基己酯、三羟甲基丙烷辛酸酯、三羟甲基丙烷壬酸酯、三羟甲基丙烷三庚酸酯、季戊四醇-2-乙基己酸酯、季戊四醇壬酸酯、季戊四醇四庚酸酯等作为酯。
具体而言,从与后述共聚物(B)的相容性的观点考虑,构成酯的醇部位优选羟基为2官能以上的醇,脂肪酸部位优选碳原子数为8以上的脂肪酸。但是,关于脂肪酸,从制造成本方面考虑,在工业上容易获得的碳原子数为20以下的脂肪酸是优选的。无论构成酯的脂肪酸是1种脂肪酸、还是2种以上的酸混合物,均可充分发挥本发明中公开的性能。作为上述酯,更具体而言,可举出三羟甲基丙烷月桂酸硬脂酸混合三酯、己二酸二异癸酯等,它们从共聚物(B)之类的饱和烃成分、与后述的具有极性基团的抗氧化剂、防蚀剂、抗磨剂、摩擦调节剂、倾点下降剂、防锈剂及消泡剂等稳定剂的相容性方面考虑是优选的。
对于本发明的润滑油组合物而言,在使用合成烃油作为润滑油基础油(A)的情况下,在将润滑油组合物整体作为100质量%时,优选以5~20质量%的量包含脂肪酸酯。通过含有5质量%以上的脂肪酸酯,可得到对于各种内燃机、工业机械内部的树脂、弹性体之类的润滑油密封材料的良好的适应性。具体而言,可抑制润滑油密封材料的溶胀。从氧化稳定性或耐热性的观点考虑,酯的量优选为20质量%以下。润滑油组合物中包含矿物油时,矿物油自身具有抑制润滑油密封材料溶胀的效果,因此并非必须使用脂肪酸酯。
本发明的润滑油组合物中,作为润滑油基础油(A),可单独使用1种矿物油系润滑油基础油或合成系润滑油基础油,另外,也可使用选自矿物油系润滑油基础油、合成系润滑油基础油中的2种以上润滑油的任意混合物等。
在按照JIS K2283中记载的方法进行测定的情况下,润滑油基础油(A)在100℃时的运动粘度为1~10mm2/s,优选为2~7mm2/s。运动粘度比上述范围高时,润滑油组合物的温度粘度特性差,运动粘度比上述范围低时,润滑油组合物在高温下的蒸发失重增加。
<(B)乙烯-α-烯烃共聚物>
乙烯-α-烯烃共聚物(B)为具有下述(B1)、(B2)、(B3)及(B4)的特性的、乙烯与α-烯烃的共聚物。
(B1)分子量
对于乙烯-α-烯烃共聚物(B)而言,利用凝胶渗透色谱法(GPC)按照后述的方法测定、并按照标准聚苯乙烯换算得到的峰顶分子量为3,000~10,000,优选为5,000~9,000,进一步优选为6,000~8,000。此处,峰顶分子量是指形成分子量分布曲线中的dw/dLog(M)(M为分子量,w为具有对应的分子量的成分的重量分数。)的最高的极大值的分子量。该分子量存在多个时,将较大的分子量作为峰顶分子量。峰顶分子量低于上述范围时,后述的润滑油组合物的粘度温度特性、以及低温粘度特性恶化,高于上述范围时,润滑油组合物的剪切稳定性恶化。
需要说明的是,本说明书中,“分子量分布曲线”或“GPC图谱”是指微分分子量分布曲线。
(B2)熔点
乙烯-α-烯烃共聚物(B)不具有利用差热量热计(DSC)测得的熔融峰。所谓不具有熔融峰,是指在DSC测定中实质上未观测到熔化热ΔH,共聚物不具有熔点,即,是指共聚物为非晶质的聚合物。所谓实质上未测量到熔化热(ΔH),是指在DSC测定中未观测到峰,或观测到的熔化热为1J/g以下。乙烯-α-烯烃共聚物具有结晶性时,润滑油组合物的低温粘度特性恶化。DSC的测定条件如实施例中所记载。
(B3)B值
对于乙烯-α-烯烃共聚物(B)而言,下述式[1]表示的B值为1.1以上,优选为1.2以上。
[数学式2]
式[1]中,PE表示乙烯成分所占的摩尔分数,PO表示α-烯烃成分所占的摩尔分数,POE表示全部二单元组链的乙烯-α-烯烃链的摩尔分数。
上述B值越大,表示嵌段的链越少,为乙烯及α-烯烃的分布一致、组成分布窄的共聚物。该嵌段的链的长度对共聚物的物性方面的特性造成影响,B值越大,则嵌段的链越短、倾点(pour point)越低,显示良好的低温粘度特性。
B值是表示共聚物中的共聚单体链分布的无规性的指标,上述式[1]中的PE、PO及POE可通过测定13C-NMR波谱、基于J.C.Randall[Macromolecules,15,353(1982)]、J.Ray[Macromolecules,10,773(1977)]等的报道而求出。
B值的测定条件如实施例中所记载。
(B4)100℃运动粘度
对于乙烯-α-烯烃共聚物(B)而言,利用JIS K2283中记载的方法测得的100℃时的运动粘度为140~500mm2/s,优选为250~450mm2/s,更优选为250~380mm2/s。乙烯-α-烯烃共聚物(B)在100℃时的运动粘度在上述范围内时,从润滑油组合物的低温粘度特性方面考虑是优选的。
对于乙烯-α-烯烃共聚物(B)而言,乙烯含量通常在30~70摩尔%的范围内,优选在40~70摩尔%的范围内,特别优选在45~65摩尔%的范围内。低于上述范围时,粘度温度特性恶化,高于上述范围时,存在由于分子内的乙烯链伸长而呈现出结晶性的情况,低温粘度特性恶化。
需要说明的是,乙烯含量可按照“高分子分析手册”(朝仓书店发行,P163~170)中记载的方法利用13C-NMR测定。另外,也可将已利用该方法求出乙烯含量的试样作为已知试样,使用傅里叶变换红外光谱(FT-IR)进行测定。
此外,对于乙烯-α-烯烃共聚物(B)而言,利用1H-NMR测得的来源于乙烯基、亚乙烯基(vinylidene)、二取代烯烃及三取代烯烃的分子链双键的总个数相对于1000个碳原子而言小于0.5个,优选小于0.3个,更优选小于0.2个,进一步优选小于0.1个。分子链双键量在该范围内时,润滑油组合物的耐热性变得良好。
作为乙烯-α-烯烃共聚物(B)中使用的α-烯烃,可例举丙烯、1-丁烯、1-戊烯、3-甲基-1-丁烯、1-己烯、4-甲基-1-戊烯、3-甲基-1-戊烯、1-辛烯、1-癸烯、1-十二碳烯、1-十四碳烯、1-十六碳烯、1-十八碳烯、1-二十碳烯、乙烯基环己烷等碳原子数为3~20的直链状或支链状的α-烯烃。作为α-烯烃,优选为碳原子数为3~10的直链状或支链状的α-烯烃,更优选为丙烯、1-丁烯、1-己烯及1-辛烯,从使用了得到的共聚物的润滑油组合物的剪切稳定性方面考虑,最优选为丙烯。这些α-烯烃可单独使用1种,或者组合使用2种以上。
另外,也可使选自含有极性基团的单体、芳香族乙烯基化合物、及环状烯烃中的至少1种在反应体系中共存而进行聚合。相对于乙烯和碳原子数为3~20的α-烯烃的合计100质量份,可以以例如20质量份以下、优选为10质量份以下的量使用其他单体。
作为含有极性基团的单体,可例举丙烯酸、甲基丙烯酸、富马酸、马来酸酐等α,β-不饱和羧酸类;及它们的钠盐等金属盐类;丙烯酸甲酯、丙烯酸乙酯、丙烯酸正丙酯、甲基丙烯酸甲酯、甲基丙烯酸乙酯等α,β-不饱和羧酸酯类;乙酸乙烯酯、丙酸乙烯酯等乙烯基酯类;丙烯酸缩水甘油酯、甲基丙烯酸缩水甘油酯等不饱和缩水甘油基化合物类等。
作为芳香族乙烯基化合物,可例举苯乙烯、邻甲基苯乙烯、间甲基苯乙烯、对甲基苯乙烯、邻,对-二甲基苯乙烯、甲氧基苯乙烯、乙烯基苯甲酸、乙烯基苯甲酸甲酯、乙酸乙烯基苄酯、羟基苯乙烯、对氯苯乙烯、二乙烯基苯、α-甲基苯乙烯、烯丙基苯等。
作为环状烯烃,可例举环戊烯、环庚烯、降冰片烯、5-甲基-2-降冰片烯、四环十二碳烯等碳原子数为3~30、优选为3~20的环状烯烃类。
作为乙烯-α-烯烃共聚物(B)的制造方法,没有特别限制,可举出专利文献5及专利文献6中记载那样的利用包含钒化合物和有机铝化合物的钒系催化剂的方法。另外,作为以高聚合活性制造共聚物的方法,可使用专利文献7~9中记载那样的使用包含二茂锆等茂金属化合物和有机铝氧化合物(铝氧烷)的催化剂体系的方法等,这些方法由于可降低得到的共聚物的氯含量、及丙烯的2,1-插入,因而是更优选的。与使用茂金属系催化剂的方法相比,利用钒系催化剂的方法在助催化剂中更多地使用氯化合物,因此,有可能在得到的乙烯-α-烯烃共聚物(B)中残留微量的氯。
另一方面,对于使用茂金属系催化剂的方法而言,由于实质上未残留氯,因而无需考虑内燃机、机械等中的金属部分的腐蚀的可能性。另外,丙烯的2,1-插入降低可进一步减少共聚物分子内的乙烯链,可提高粘度温度特性、低温粘度特性。
尤其是通过使用以下这样的方法,可得到在分子量控制、分子量分布、非晶性、B值方面具有良好的性能均衡性的乙烯-α-烯烃共聚物(B)。
乙烯-α-烯烃共聚物(B)可通过下述方式制造:在包含下述通式[I]表示的交联茂金属化合物(a)以及化合物(b)的烯烃聚合催化剂的存在下,使乙烯与碳原子数为3~20的α-烯烃共聚,所述化合物(b)为选自由有机金属化合物(b-1)、有机铝氧化合物(b-2)及能与上述交联茂金属化合物(a)反应而形成离子对的化合物(b-3)组成的组中的至少1种。
[化学式1]
<交联茂金属化合物>
交联茂金属化合物(a)由上述式[I]表示。上述式[I]表示的交联茂金属化合物形成嵌段的链短、即B值大的共聚物。以下说明式[I]中的Y、M、R1~R14、Q、n及j。
(Y、M、R1~R12、Q、n及j)
Y为第14族原子,例如,可举出碳原子、硅原子、锗原子及锡原子,优选为碳原子或硅原子,更优选为碳原子。
M为钛原子、锆原子或铪原子,优选为锆原子。
R1~R12为选自由氢原子、碳原子数为1~20的烃基、含硅基团、含氮基团、含氧基团、卤素原子及含卤素基团组成的组中的原子或取代基,各自可以相同也可以不同。另外,R1~R12中的相邻的取代基可以相互键合而形成环,也可不相互键合。
此处,作为碳原子数为1~20的烃基,可例举碳原子数为1~20的烷基、碳原子数为3~20的环状饱和烃基、碳原子数为2~20的链状不饱和烃基、碳原子数为3~20的环状不饱和烃基、碳原子数为1~20的亚烷基、碳原子数为6~20的亚芳基等。
作为碳原子数为1~20的烷基,可例举作为直链状饱和烃基的甲基、乙基、正丙基、烯丙(allyl)基、正丁基、正戊基、正己基、正庚基、正辛基、正壬基、正癸基等;作为支链状饱和烃基的异丙基、异丁基、仲丁基、叔丁基、叔戊基、新戊基、3-甲基戊基、1,1-二乙基丙基、1,1-二甲基丁基、1-甲基-1-丙基丁基、1,1-丙基丁基、1,1-二甲基-2-甲基丙基、1-甲基-1-异丙基-2-甲基丙基、环丙基甲基等。烷基的碳原子数优选为1~6。
作为碳原子数为3~20的环状饱和烃基,可例举作为环状饱和烃基的环丙基、环丁基、环戊基、环己基、环庚基、环辛基、降冰片烯基(norbornenyl group)、1-金刚烷基、2-金刚烷基等;作为环状饱和烃基的氢原子被碳原子数为1~17的烃基取代而成的基团的3-甲基环戊基、3-甲基环己基、4-甲基环己基、4-环己基环己基、4-苯基环己基等。环状饱和烃基的碳原子数优选为5~11。
作为碳原子数为2~20的链状不饱和烃基,可例举作为链烯基的乙烯基(vinyl group)、1-丙烯基、2-丙烯基(烯丙基)、1-甲基乙烯基(异丙烯基)等;作为炔基的乙炔基、1-丙炔基、2-丙炔基(炔丙基)等。链状不饱和烃基的碳原子数优选为2~4。
作为碳原子数为3~20的环状不饱和烃基,可例举作为环状不饱和烃基的环戊二烯基、降冰片基、苯基、萘基、茚基、薁基、菲基、蒽基等;作为环状不饱和烃基的氢原子被碳原子数为1~15的烃基取代而成的基团的3-甲基苯基(间甲苯基)、4-甲基苯基(对甲苯基)、4-乙基苯基、4-叔丁基苯基、4-环己基苯基、联苯基、3,4-二甲基苯基、3,5-二甲基苯基、2,4,6-三甲基苯基(均三甲苯基(mesityl group))等;作为直链状烃基或支链状饱和烃基的氢原子被碳原子数为3~19的环状饱和烃基或环状不饱和烃基取代而成的基团的苄基、枯基等。环状不饱和烃基的碳原子数优选为6~10。
作为碳原子数为1~20的亚烷基,可例举亚甲基、亚乙基、二甲基亚甲基(异亚丙基(isopropylidene))、乙基亚甲基、甲基亚乙基、亚正丙基等。亚烷基的碳原子数优选为1~6。
作为碳原子数为6~20的亚芳基,可例举邻亚苯基、间亚苯基、对亚苯基、4,4’-亚联苯基(biphenylylene group)等。亚芳基的碳原子数优选为6~12。
作为含硅基团,可例举作为碳原子数为1~20的烃基中碳原子被硅原子取代而成的基团的三甲基甲硅烷基、三乙基甲硅烷基、叔丁基二甲基甲硅烷基、三异丙基甲硅烷基等烷基甲硅烷基;二甲基苯基甲硅烷基、甲基二苯基甲硅烷基、叔丁基二苯基甲硅烷基等芳基甲硅烷基;五甲基乙硅烷基;三甲基甲硅烷基甲基等。烷基甲硅烷基的碳原子数优选为1~10,芳基甲硅烷基的碳原子数优选为6~18。
作为含氮基团,可例举氨基、作为上述的碳原子数为1~20的烃基或含硅基团中=CH-结构单元被氮原子取代而成的基团、-CH2-结构单元被键合有碳原子数为1~20的烃基的氮原子取代而成的基团、或-CH3结构单元被键合有碳原子数为1~20的烃基的氮原子或腈基取代而成的基团的二甲基氨基、二乙基氨基、N-吗啉基、二甲基氨基甲基、氰基、吡咯烷基、哌啶基、吡啶基等;N-吗啉基及硝基等。作为含氮基团,优选为二甲基氨基、N-吗啉基。
作为含氧基团,可例举羟基、作为上述的碳原子数为1~20的烃基、含硅基团或含氮基团中-CH2-结构单元被氧原子或羰基取代而成的基团、或-CH3结构单元被键合有碳原子数为1~20的烃基的氧原子取代而成的基团的甲氧基、乙氧基、叔丁氧基、苯氧基、三甲基甲硅烷氧基、甲氧基乙氧基、羟基甲基、甲氧基甲基、乙氧基甲基、叔丁氧基甲基、1-羟基乙基、1-甲氧基乙基、1-乙氧基乙基、2-羟基乙基、2-甲氧基乙基、2-乙氧基乙基、2-氧杂亚正丁基、2-氧杂亚正戊基、3-氧杂亚正戊基、醛基、乙酰基、丙酰基、苯甲酰基、三甲基甲硅烷基羰基、氨基甲酰基、甲基氨基羰基、羧基、甲氧基羰基、羧甲基、乙氧基羧甲基、氨基甲酰甲基、呋喃基、吡喃基等。作为含氧基团,优选甲氧基。
作为卤素原子,可例举作为第17族元素的氟、氯、溴、碘等。
作为含卤素基团,可例举在上述的碳原子数为1~20的烃基、含硅基团、含氮基团或含氧基团中氢原子被卤素原子取代而得到的基团、即三氟甲基、三溴甲基、五氟乙基、五氟苯基等。
Q选自卤素原子、碳原子数为1~20的烃基、阴离子配体及可利用孤对电子配位的中性配体,可以是相同的,或者为不同的组合。
卤素原子及碳原子数为1~20的烃基的详细情况如上文所述。Q为卤素原子时,优选氯原子。Q为碳原子数为1~20的烃基时,该烃基的碳原子数优选为1~7。
作为阴离子配体,可例举甲氧基、叔丁氧基、苯氧基等烷氧基;乙酸根、苯甲酸根等羧酸根基团(carboxylate group);甲磺酸根、甲苯磺酸根等磺酸根基团(sulfonate group)等。
作为可利用孤对电子配位的中性配体,可例举三甲基膦、三乙基膦、三苯基膦、二苯基甲基膦等有机磷化合物;四氢呋喃、乙醚、二氧杂环己烷、1,2-二甲氧基乙烷等醚化合物等。
j为1~4的整数,优选为2。
n为1~4的整数,优选为1或2,进一步优选为1。
R13及R14为选自由氢原子、碳原子数为1~20的烃基、芳基、取代芳基、含硅基团、含氮基团、含氧基团、卤素原子及含卤素基团组成的组中的原子或取代基,各自可以相同也可以不同。另外,R13及R14可以相互键合而形成环,也可不相互键合。
关于碳原子数为1~20的烃基、含硅基团、含氮基团、含氧基团、卤素原子及含卤素基团的详细情况,如上文所述。
作为芳基,与上述的碳原子数为3~20的环状不饱和烃基的例子有一部分重复,可例举作为由芳香族化合物衍生的取代基的苯基、1-萘基、2-萘基、蒽基、菲基、并四苯基(tetracenyl group)、基(chrysenyl group)、芘基、茚基、薁基、吡咯基、吡啶基、呋喃基、噻吩基等。作为芳基,优选苯基或2-萘基。
作为上述芳香族化合物,可例举作为芳香族烃及杂环式芳香族化合物的苯、萘、蒽、菲、并四苯(tetracene)、芘、茚、薁、呲咯、呲啶、呋喃、噻吩等。
作为取代芳基,与上述的碳原子数为3~20的环状不饱和烃基的例子有一部分重复,可举出上述芳基所具有的1个以上的氢原子被选自由碳原子数为1~20的烃基、芳基、含硅基团、含氮基团、含氧基团、卤素原子及含卤素基团组成的组中的至少1种取代基取代而成的基团,具体而言,可例举3-甲基苯基(间甲苯基)、4-甲基苯基(对甲苯基)、3-乙基苯基、4-乙基苯基、3,4-二甲基苯基、3,5-二甲基苯基、联苯基、4-(三甲基甲硅烷基)苯基、4-氨基苯基、4-(二甲基氨基)苯基、4-(二乙基氨基)苯基、4-吗啉基苯基、4-甲氧基苯基、4-乙氧基苯基、4-苯氧基苯基、3,4-二甲氧基苯基、3,5-二甲氧基苯基、3-甲基-4-甲氧基苯基、3,5-二甲基-4-甲氧基苯基、3-(三氟甲基)苯基、4-(三氟甲基)苯基、3-氯苯基、4-氯苯基、3-氟苯基、4-氟苯基、5-甲基萘基、2-(6-甲基)吡啶基等。
上述式[I]表示的交联茂金属化合物(a)中,n优选为1。这样的交联茂金属化合物(以下,也称为“交联茂金属化合物(a-1)”。)由下述通式[II]表示。
[化学式2]
式[II]中,Y、M、R1、R2、R3、R4、R5、R6、R7、R8、R9、R10、R11、R12、R13、R14、Q及j的定义等如上文所述。
与上述式[I]中的n为2~4的整数的化合物相比,交联茂金属化合物(a-1)的制造工序简单、制造成本降低,进而,通过使用该交联茂金属化合物(a-1),可获得降低乙烯-α-烯烃共聚物(B)的制造成本这样的优点。
上述式[II]表示的交联茂金属化合物(a-1)中,优选R1、R2、R3及R4均为氢。这样的交联茂金属化合物(以下,也称为“交联茂金属化合物(a-2)”。)由下述通式[III]表示。
[化学式3]
式[III]中,Y、M、R5、R6、R7、R8、R9、R10、R11、R12、R13、R14、Q及j的定义等如上文所述。
与上述式[I]中的R1、R2、R3及R4中的任意一个以上被氢原子以外的取代基取代而成的化合物相比,交联茂金属化合物(a-2)的制造工序简单、制造成本降低,进而,通过使用该交联茂金属化合物(a-2),可获得降低乙烯-α-烯烃共聚物(B)的制造成本这样的优点。另外,虽然已知通常进行高温聚合会导致乙烯-α-烯烃共聚物(B)的无规性降低,但在包含该交联茂金属化合物(a-2)的烯烃聚合催化剂的存在下将乙烯与选自碳原子数为3~20的α-烯烃中的1种以上的单体共聚时,还可获得以下这样的优点:即使是高温聚合,得到的乙烯-α-烯烃共聚物(B)的无规性也高。
上述式[III]表示的交联茂金属化合物(a-2)中,优选R13及R14中的任一者为芳基或取代芳基。这样的交联茂金属化合物(a-3)与R13及R14均为芳基及取代芳基以外的取代基的情况相比,可获得生成的乙烯-α-烯烃共聚物(B)中的双键量少这样的优点。
交联茂金属化合物(a-3)中,进一步优选的是,R13及R14中的任一者为芳基或取代芳基,另一者为碳原子数为1~20的烷基,特别优选的是,R13及R14中的任一者为芳基或取代芳基,另一者为甲基。这样的交联茂金属化合物(以下,也称为“交联茂金属化合物(a-4)”。)与R13及R14均为芳基或取代芳基的情况相比,可获得这样的优点:生成的乙烯-α-烯烃共聚物(B)中的双键量与聚合活性的均衡性优异,通过使用该交联茂金属化合物,可降低乙烯-α-烯烃共聚物(B)的制造成本。
在某一恒定的聚合器内总压力及温度的条件下实施聚合时,产生以下这样的问题:由导入氢而导致的氢分压的上升引起作为聚合单体的烯烃的分压降低,尤其是在氢分压高的区域,聚合速度降低。聚合反应器的在其设计上被允许的内部总压力受到限制,因此,尤其是在制造低分子量的烯烃聚合物时,若需要过度地导入氢,则烯烃分压显著降低,因而聚合活性有时降低。然而,使用交联茂金属化合物(a-4)制造乙烯-α-烯烃共聚物(B)时,与使用上述交联茂金属化合物(a-3)的情况相比,可获得以下这样的优点:导入到聚合反应器中的氢量降低,聚合活性提高,可降低乙烯-α-烯烃共聚物(B)的制造成本。
上述交联茂金属化合物(a-4)中,R6及R11优选为可与相邻的取代基相互键合而形成环的碳原子数为1~20的烷基及碳原子数为1~20的亚烷基。这样的交联茂金属化合物(以下,也称为“交联茂金属化合物(a-5)”。)与R6及R11被碳原子数为1~20的烷基及碳原子数为1~20的亚烷基以外的取代基取代而成的化合物相比,制造工序简单,制造成本降低,进而,通过使用该交联茂金属化合物(a-5),可获得降低乙烯-α-烯烃共聚物(B)的制造成本这样的优点。
上述通式[I]表示的交联茂金属化合物(a)、上述通式[II]表示的交联茂金属化合物(a-1)、上述通式[III]表示的交联茂金属化合物(a-2)、以及上述交联茂金属化合物(a-3)、(a-4)及(a-5)中,M进一步优选为锆原子。在包含M为锆原子的上述交联茂金属化合物的烯烃聚合催化剂的存在下将乙烯与选自碳原子数为3~20的α-烯烃中的1种以上的单体共聚时,与M为钛原子或铪原子的情况相比,可获得以下这样的优点:聚合活性高,可降低乙烯-α-烯烃共聚物(B)的制造成本。
作为这样的交联茂金属化合物(a),可举出:
[二甲基亚甲基(η5-环戊二烯基)(η5-芴基)]二氯化锆、[二甲基亚甲基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆、[二甲基亚甲基(η5-环戊二烯基)(η5-3,6-二叔丁基芴基)]二氯化锆、[二甲基亚甲基(η5-环戊二烯基)(η5-八甲基八氢二苯并芴基)]二氯化锆、[二甲基亚甲基(η5-环戊二烯基)(η5-四甲基八氢二苯并芴基)]二氯化锆,
[亚环己基(η5-环戊二烯基)(η5-芴基)]二氯化锆、[亚环己基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆、[亚环己基(η5-环戊二烯基)(η5-3,6-二叔丁基芴基)]二氯化锆、[亚环己基(η5-环戊二烯基)(η5-八甲基八氢二苯并芴基)]二氯化锆、[亚环己基(η5-环戊二烯基)(η5-四甲基八氢二苯并芴基)]二氯化锆,
[二苯基亚甲基(η5-环戊二烯基)(η5-芴基)]二氯化锆、[二苯基亚甲基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆、二苯基亚甲基(η5-2-甲基-4-叔丁基环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆、[二苯基亚甲基(η5-环戊二烯基)(η5-3,6-二叔丁基芴基)]二氯化锆、[二苯基亚甲基(η5-环戊二烯基)(η5-八甲基八氢二苯并芴基)]二氯化锆、[二苯基亚甲基(η5-环戊二烯基)(η5-四甲基八氢二苯并芴基)]二氯化锆,
[甲基苯基亚甲基(η5-环戊二烯基)(η5-芴基)]二氯化锆、[甲基苯基亚甲基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆、[甲基苯基亚甲基(η5-环戊二烯基)(η5-3,6-二叔丁基芴基)]二氯化锆、[甲基苯基亚甲基(η5-环戊二烯基)(η5-八甲基八氢二苯并芴基)]二氯化锆、[甲基苯基亚甲基(η5-环戊二烯基)(η5-四甲基八氢二苯并芴基)]二氯化锆,
[甲基(3-甲基苯基)亚甲基(η5-环戊二烯基)(η5-芴基)]二氯化锆、[甲基(3-甲基苯基)亚甲基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆、[甲基(3-甲基苯基)亚甲基(η5-环戊二烯基)(η5-3,6-二叔丁基芴基)]二氯化锆、[甲基(3-甲基苯基)亚甲基(η5-环戊二烯基)(η5-八甲基八氢二苯并芴基)]二氯化锆、[甲基(3-甲基苯基)亚甲基(η5-环戊二烯基)(η5-四甲基八氢二苯并芴基)]二氯化锆,
[二苯基亚甲硅基(η5-环戊二烯基)(η5-芴基)]二氯化锆、[二苯基亚甲硅基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆,[二苯基亚甲硅基(η5-环戊二烯基)(η5-3,6-二叔丁基芴基)]二氯化锆、[二苯基亚甲硅基(η5-环戊二烯基)(η5-八甲基八氢二苯并芴基)]二氯化锆、[二苯基亚甲硅基(η5-环戊二烯基)(η5-四甲基八氢二苯并芴基)]二氯化锆,
[双(3-甲基苯基)亚甲硅基(η5-环戊二烯基)(η5-芴基)]二氯化锆、[双(3-甲基苯基)亚甲硅基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆、[双(3-甲基苯基)亚甲硅基(η5-环戊二烯基)(η5-3,6-二叔丁基芴基)]二氯化锆、[双(3-甲基苯基)亚甲硅基(η5-环戊二烯基)(η5-八甲基八氢二苯并芴基)]二氯化锆、[双(3-甲基苯基)亚甲硅基(η5-环戊二烯基)(η5-四甲基八氢二苯并芴基)]二氯化锆,
[二环己基亚甲硅基(η5-环戊二烯基)(η5-芴基)]二氯化锆、[二环己基亚甲硅基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆,[二环己基亚甲硅基(η5-环戊二烯基)(η5-3,6-二叔丁基芴基)]二氯化锆、[二环己基亚甲硅基(η5-环戊二烯基)(η5-八甲基八氢二苯并芴基)]二氯化锆、[二环己基亚甲硅基(η5-环戊二烯基)(η5-四甲基八氢二苯并芴基)]二氯化锆,
[亚乙基(η5-环戊二烯基)(η5-芴基)]二氯化锆、[亚乙基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆、[亚乙基(η5-环戊二烯基)(η5-3,6-二叔丁基芴基)]二氯化锆、[亚乙基(η5-环戊二烯基)(η5-八甲基八氢二苯并芴基)]二氯化锆、[亚乙基(η5-环戊二烯基)(η5-四甲基八氢二苯并芴基)]二氯化锆,等等。
可例举将这些化合物的锆原子替换为铪原子而得到的化合物或将氯配体替换为甲基而得到的化合物等,但交联茂金属化合物(a)不限于上述示例。需要说明的是,分别地,作为例举的交联茂金属化合物(a)的构成部分的η5-四甲基八氢二苯并芴基表示4,4,7,7-四甲基-(5a,5b,11a,12,12a-η5)-1,2,3,4,7,8,9,10-八氢二苯并[b,H]芴基,η5-八甲基八氢二苯并芴基表示1,1,4,4,7,7,10,10-八甲基-(5a,5b,11a,12,12a-η5)-1,2,3,4,7,8,9,10-八氢二苯并[b,H]芴基。
<化合物(b)>
本发明中使用的聚合催化剂包含:上述的交联茂金属化合物(a)、以及选自由有机金属化合物(b-1)、有机铝氧化合物(b-2)及能与交联茂金属化合物(a)反应而形成离子对的化合物(b-3)组成的组中的至少1种化合物(b)。
作为有机金属化合物(b-1),具体而言,可使用下述这样的元素周期表第1、2族及第12、13族的有机金属化合物。
(b-1a)通式RamAl(ORb)nHpXq表示的有机铝化合物。
(式中,Ra及Rb相互可以相同也可以不同,表示碳原子数为1~15、优选为1~4的烃基,X表示卤素原子,m为0<m≤3的数,n为0≤n<3的数,p为0≤p<3的数,q为0≤q<3的数,并且m+n+p+q=3)
作为这样的化合物,可例举:
三甲基铝、三乙基铝、三正丁基铝、三正己基铝、三正辛基铝等三正烷基铝;三异丙基铝、三异丁基铝、三仲丁基铝、三叔丁基铝、三-2-甲基丁基铝、三-3-甲基己基铝、三-2-乙基己基铝等三支链状烷基铝;三环己基铝、三环辛基铝等三环烷基铝;三苯基铝、三(4-甲基苯基)铝等三芳基铝;二异丙基氢化铝、二异丁基氢化铝等二烷基氢化铝;通式(i-C4H9)xAly(C5H10)z(式中,x、y、z为正数,z≤2x。)表示的异戊二烯基铝等链烯基铝;异丁基甲醇铝、异丁基乙醇铝等烷基烷醇铝;二甲基甲醇铝、二乙基乙醇铝、二丁基丁醇铝等二烷基烷醇铝;乙基倍半乙醇铝、丁基倍半丁醇铝等烷基倍半烷醇铝;具有通式Ra2.5Al(ORb)0.5等表示的平均组成的被部分烷氧基化的烷基铝;二乙基苯酚铝、二乙基(2,6-二叔丁基-4-甲基苯酚)铝等烷基芳氧基铝;二甲基氯化铝、二乙基氯化铝、二丁基氯化铝、二乙基溴化铝、二异丁基氯化铝等二烷基卤化铝;乙基倍半氯化铝、丁基倍半氯化铝、乙基倍半溴化铝等烷基倍半卤化铝;乙基二氯化铝等烷基二卤化铝等被部分卤化的烷基铝;二乙基氢化铝、二丁基氢化铝等二烷基氢化铝;乙基二氢化铝、丙基二氢化铝等烷基二氢化铝及其他的被部分氢化的烷基铝;乙基乙氧基氯化铝、丁基丁氧基氯化铝、乙基乙氧基溴化铝等被部分烷氧基化及卤化的烷基铝等。另外,也可使用与上述通式RamAl(ORb)nHpXq表示的化合物类似的化合物,例如,可举出2个以上的铝化合物介由氮原子键合而成的有机铝化合物。作为这样的化合物,具体而言,可举出(C2H5)2AlN(C2H5)Al(C2H5)2等。
(b-1b)通式M2AlRa4表示的元素周期表第1族金属与铝的烷基络合物。(式中,M2表示Li、Na或K,Ra表示碳原子数为1~15、优选为1~4的烃基。)
作为这样的化合物,可例举LiAl(C2H5)4、LiAl(C7H15)4等。
(b-1c)通式RaRbM3表示的元素周期表第2族或第12族金属的二烷基化合物。(式中,Ra及Rb相互可以相同也可以不同,表示碳原子数为1~15、优选为1~4的烃基,M3为Mg、Zn或Cd。)
作为有机铝氧化合物(b-2),可直接使用现有已知的铝氧烷。具体而言,可举出下述通式[IV]表示的化合物及下述通式[V]表示的化合物。
[化学式4]
式[IV]及[V]中,R表示碳原子数为1~10的烃基,n表示2以上的整数。
尤其是,可利用R为甲基、n为3以上、优选为10以上的甲基铝氧烷。也可在这些铝氧烷类中混入若干有机铝化合物。
本发明中,在高温下进行乙烯与碳原子数为3以上的α-烯烃的共聚时,也可应用日本特开平2-78687号公报中例举的那样的不溶于苯的有机铝氧化合物。另外,也可合适地利用日本特开平2-167305号公报中记载的有机铝氧化合物、日本特开平2-24701号公报、日本特开平3-103407号公报中记载的具有两种以上的烷基的铝氧烷等。需要说明的是,可用于本发明中的“不溶于苯的有机铝氧化合物”是指,溶解于60℃的苯中的Al成分以Al原子换算计通常为10%以下、优选为5%以下、特别优选为2%以下的、相对于苯为不溶性或难溶性的化合物。
另外,作为有机铝氧化合物(b-2),也可举出下述通式[VI]表示的那样的修饰甲基铝氧烷等。
[化学式5]
式[VI]中,R表示碳原子数为1~10的烃基,m及n各自独立地表示2以上的整数。
该修饰甲基铝氧烷为使用三甲基铝和三甲基铝以外的烷基铝制备的化合物。这样的化合物通常被称为MMAO。这样的MMAO可利用美国专利4960878号说明书及美国专利5041584号说明书中列举的方法制备。另外,使用三甲基铝和三异丁基铝制备的R为异丁基的物质也由Tosoh Finechem Corporation等以MMAO、TMAO这样的名称在市场上销售。这样的MMAO为改良了在各种溶剂中的溶解性及保存稳定性的铝氧烷,具体而言,与上述式[IV]表示的化合物及[V]表示的化合物中的相对于苯为不溶性或难溶性的化合物不同,溶解于脂肪族烃、脂环族烃。
此外,作为有机铝氧化合物(b-2),还可举出下述通式[VII]表示的包含硼的有机铝氧化合物。
[化学式6]
式[VII]中,Rc表示碳原子数为1~10的烃基。Rd相互可以相同也可以不同,表示氢原子、卤素原子或碳原子数为1~10的烃基。
作为能与交联茂金属化合物(a)反应而形成离子对的化合物(b-3)(以下,有时简称为“离子化离子性化合物”或仅简称为“离子性化合物”。),可举出日本特开平1-501950号公报、日本特开平1-502036号公报、日本特开平3-179005号公报、日本特开平3-179006号公报、日本特开平3-207703号公报、日本特开平3-207704号公报、美国专利5321106号说明书等中记载的路易斯酸、离子性化合物、硼烷化合物及碳硼烷化合物等。此外,还可举出杂多化合物及同多化合物。
本发明中优选使用的离子化离子性化合物为下述通式[VIII]表示的硼化合物。
[化学式7]
式[VIII]中,作为Re+,可举出H+、碳鎓阳离子、氧鎓阳离子、铵阳离子、鏻阳离子、环庚基三烯基阳离子(cycloheptyltrienyl cation)、具有过渡金属的二茂铁鎓阳离子等。Rf~Ri相互可以相同也可以不同,为选自碳原子数为1~20的烃基、含硅基团、含氮基团、含氧基团、卤素原子及含卤素基团中的取代基,优选为取代芳基。
作为上述碳鎓阳离子,具体而言,可举出三苯基碳鎓阳离子、三(4-甲基苯基)碳鎓阳离子、三(3,5-二甲基苯基)碳鎓阳离子等三取代碳鎓阳离子等。
作为上述铵阳离子,具体而言,可举出三甲基铵阳离子、三乙基铵阳离子、三(正丙基)铵阳离子、三异丙基铵阳离子、三(正丁基)铵阳离子、三异丁基铵阳离子等三烷基取代铵阳离子;N,N-二甲基苯胺鎓阳离子、N,N-二乙基苯胺鎓阳离子、N,N-2,4,6-五甲基苯胺鎓阳离子等N,N-二烷基苯胺鎓阳离子;二异丙基铵阳离子、二环己基铵阳离子等二烷基铵阳离子等。
作为上述鏻阳离子,具体而言,可举出三苯基鏻阳离子、三(4-甲基苯基)鏻阳离子、三(3,5-二甲基苯基)鏻阳离子等三芳基鏻阳离子等。
作为Re+,上述具体例中,优选碳鎓阳离子、铵阳离子等,特别优选三苯基碳鎓阳离子、N,N-二甲基苯胺鎓阳离子、N,N-二乙基苯胺鎓阳离子。
本发明中优选使用的离子化离子性化合物中,作为包含碳鎓阳离子的化合物,可例举四苯基硼酸三苯基碳鎓、四(五氟苯基)硼酸三苯基碳鎓、四{3,5-二-(三氟甲基)苯基}硼酸三苯基碳鎓、四(五氟苯基)硼酸三(4-甲基苯基)碳鎓、四(五氟苯基)硼酸三(3,5-二甲基苯基)碳鎓等。
本发明中优选使用的离子化离子性化合物中,作为包含三烷基取代铵阳离子的化合物,可例举四苯基硼酸三乙基铵、四苯基硼酸三丙基铵、四苯基硼酸三(正丁基)铵、四(4-甲基苯基)硼酸三甲基铵、四(2-甲基苯基)硼酸三甲基铵、四(五氟苯基)硼酸三(正丁基)铵、四(五氟苯基)硼酸三乙基铵、四(五氟苯基)硼酸三丙基铵、四(2,4-二甲基苯基)硼酸三丙基铵、四(3,5-二甲基苯基)硼酸三(正丁基)铵、四{4-(三氟甲基)苯基}硼酸三(正丁基)铵、四{3,5-二(三氟甲基)苯基}硼酸三(正丁基)铵、四(2-甲基苯基)硼酸三(正丁基)铵、四苯基硼酸双十八烷基甲基铵、四(4-甲基苯基)硼酸双十八烷基甲基铵、四(4-甲基苯基)硼酸双十八烷基甲基铵、四(五氟苯基)硼酸双十八烷基甲基铵、四(2,4-二甲基苯基)硼酸双十八烷基甲基铵、四(3,5-二甲基苯基)硼酸双十八烷基甲基铵、四{4-(三氟甲基)苯基}硼酸双十八烷基甲基铵、四{3,5-二(三氟甲基)苯基}硼酸双十八烷基甲基铵、双十八烷基甲基铵等。
本发明中优选使用的离子化离子性化合物中,作为包含N,N-二烷基苯胺鎓阳离子的化合物,可例举四苯基硼酸N,N-二甲基苯胺鎓、四(五氟苯基)硼酸N,N-二甲基苯胺鎓、四{3,5-二(三氟甲基)苯基}硼酸N,N-二甲基苯胺鎓、四苯基硼酸N,N-二乙基苯胺鎓、四(五氟苯基)硼酸N,N-二乙基苯胺鎓、四{3,5-二(三氟甲基)苯基}硼酸N,N-二乙基苯胺鎓、四苯基硼酸N,N-2,4,6-五甲基苯胺鎓、四(五氟苯基)硼酸N,N-2,4,6-五甲基苯胺鎓等。
本发明中优选使用的离子化离子性化合物中,作为包含二烷基铵阳离子的化合物,可例举四(五氟苯基)硼酸二正丙基铵、四苯基硼酸二环己基铵等。
此外,也可无限制地使用由日本特开2004-51676号公报例举的离子性化合物。
上述的离子性化合物(b-3)可单独使用1种,也可混合使用2种以上。
作为有机金属化合物(b-1),优选由于为市售品而容易获得的三甲基铝、三乙基铝及三异丁基铝。其中,特别优选操作容易的三异丁基铝。
作为有机铝氧化合物(b-2),优选由于为市售品而容易获得的甲基铝氧烷、及使用三甲基铝和三异丁基铝制备的MMAO。其中,特别优选在各种溶剂中的溶解性及保存稳定性已被改良的MMAO。
作为离子性化合物(b-3),优选为四(五氟苯基)硼酸三苯基碳鎓及四(五氟苯基)硼酸N,N-二甲基苯胺鎓,这是因为它们容易以市售品的形式获得,并且对提高聚合活性的贡献大。
作为化合物(b),特别优选为三异丁基铝与四(五氟苯基)硼酸三苯基碳鎓的组合、及三异丁基铝与四(五氟苯基)硼酸N,N-二甲基苯胺鎓的组合,这是因为它们大幅提高聚合活性。
<载体(c)>
本发明中,根据需要,可使用载体(c)作为烯烃聚合催化剂的构成成分。
本发明中可使用的载体(c)为无机或有机的化合物,为颗粒状或微粒状的固体。其中,作为无机化合物,优选多孔氧化物、无机氯化物、粘土、粘土矿物或离子交换性层状化合物。
作为多孔氧化物,具体而言,可使用SiO2、Al2O3、MgO、ZrO、TiO2、B2O3、CaO、ZnO、BaO、ThO2等;或包含它们的复合物或混合物,例如天然或合成沸石、SiO2-MgO、SiO2-Al2O3、SiO2-TiO2、SiO2-V2O5、SiO2-Cr2O3、SiO2-TiO2-MgO等。这些中,优选以SiO2及/或Al2O3为主成分的多孔氧化物。对于这样的多孔氧化物而言,根据种类及制法的不同,其性状有所不同,本发明中优选使用的载体的粒径为0.5~300μm、优选为1.0~200μm,比表面积在50~1000m2/g、优选100~700m2/g的范围内,微孔容积在0.3~3.0cm3/g的范围内。对于这样的载体而言,根据需要,可于100~1000℃、优选150~700℃进行烧成后使用。
作为无机氯化物,可使用MgCl2、MgBr2、MnCl2、MnBr2等。无机氯化物可直接使用,也可利用球磨机、振动磨进行粉碎后使用。另外,也可使用下述物质:在将无机氯化物溶解于醇等溶剂中后,利用析出剂使其以微粒状析出而得到的物质。
粘土通常以粘土矿物为主成分而构成。另外,离子交换性层状化合物为具有下述晶体结构、并且其中所含的离子能够交换的化合物,所述晶体结构为利用离子键等、所构成的面以弱结合力相互平行地层叠而成的晶体结构。大部分的粘土矿物为离子交换性层状化合物。另外,作为上述粘土、粘土矿物、离子交换性层状化合物,不限于天然产物,也可使用人工合成物。另外,作为粘土、粘土矿物或离子交换性层状化合物,可例举粘土、粘土矿物、或六方紧密堆积型(hexagonal closest packing type)、锑型、CdCl2型、CdI2型等具有层状晶体结构的离子结晶性化合物等。作为这样的粘土、粘土矿物,可举出高岭土、膨润土、木节粘土(kibushi clay)、蛙目粘土(gairome clay)、水铝英石、硅铁石、叶蜡石、云母类、蒙脱石类、蛭石、绿泥石类、坡缕石、高岭石、珍珠陶土、地开石、埃洛石等,作为离子交换性层状化合物,可举出α-Zr(HAsO4)2·H2O、α-Zr(HPO4)2、α-Zr(KPO4)2·3H2O、α-Ti(HPO4)2、α-Ti(HAsO4)2·H2O、α-Sn(HPO4)2·H2O、γ-Zr(HPO4)2、γ-Ti(HPO4)2、γ-Ti(NH4PO4)2·H2O等多价金属的结晶性酸性盐等。对本发明中使用的粘土、粘土矿物实施化学处理也是优选的。作为化学处理,除去在表面附着的杂质的表面处理、对粘土的晶体结构施加影响的处理等均可使用。作为化学处理,具体而言,可举出酸处理、碱处理、盐类处理、有机物处理等。
离子交换性层状化合物可以是利用离子交换性、使层间的交换性离子与其他的大体积的离子交换从而使得层间扩大的状态的层状化合物。这样的大体积的离子担负支撑层状结构的支柱的作用,通常被称为支柱(pillar)。另外,将如上文所述地在层状化合物的层间导入其他物质(客体化合物)称为插入(intercalation)。作为客体化合物,可举出TiCl4、ZrCl4等阳离子性无机化合物;Ti(OR)4、Zr(OR)4、PO(OR)3、B(OR)3等金属醇盐(R为烃基等);[Al13O4(OH)24]7+、[Zr4(OH)14]2+、[Fe3O(OCOCH3)6]+等金属氢氧化物离子等。上述化合物可单独使用1种或组合使用2种以上。另外,在将上述化合物插入时,也可使将Si(OR)4、Al(OR)3、Ge(OR)4等金属醇盐(R为烃基等)等水解缩聚而得到的聚合物、SiO2等胶体状无机化合物等共存。另外,作为支柱,可举出通过将上述金属氢氧化物离子插入至层间后进行加热脱水而生成的氧化物等。
这些中,优选粘土或粘土矿物,特别优选蒙脱石、蛭石、针钠钙石、带云母和合成云母。
关于作为载体(c)的有机化合物,可举出粒径在0.5~300μm的范围内的颗粒状或微粒状固体。具体而言,可例举以乙烯、丙烯、1-丁烯、4-甲基-1-戊烯等碳原子数为2~14的α-烯烃为主成分而生成的(共)聚合物或以乙烯基环己烷、苯乙烯为主成分而生成的(共)聚合物、及它们的改性物。
利用使用本说明书中公开那样的可生成嵌段的链短的乙烯-α-烯烃共聚物(B)的烯烃聚合催化剂的聚合方法,能够进行高温聚合。即,通过使用该烯烃聚合催化剂,能够抑制形成通过高温聚合而伸长的乙烯-α-烯烃共聚物(B)的嵌段的链。
溶液聚合中,包含生成的乙烯-α-烯烃共聚物(B)的聚合溶液的粘度在高温下降低,因此,与低温聚合时相比,可提高聚合器内的乙烯-α-烯烃共聚物(B)的浓度,作为结果,每一个聚合器的生产率提高。本发明中的乙烯及α-烯烃的共聚可利用溶液聚合、悬浮聚合(淤浆聚合)等液相聚合法或气相聚合法中的任意方法实施,从可如上所述地最大限度地享有本发明的效果这样的观点考虑,特别优选溶液聚合。
烯烃聚合催化剂的各成分的使用方法、添加顺序可任意选择。另外,可以使催化剂中的各成分的至少2种以上预先接触。
可以以如下这样的量使用交联茂金属化合物(a)(以下,也称为“成分(a)”。):每1升反应容积,通常为10-9~10-1摩尔,优选为10-8~10-2摩尔。
可以以如下这样的量使用有机金属化合物(b-1)(以下,也称为“成分(b-1)”。):成分(b-1)与成分(a)中的过渡金属原子(M)的摩尔比[(b-1)/M]通常为0.01~50,000,优选为0.05~10,000。
可以以如下这样的量使用有机铝氧化合物(b-2)(以下,也称为“成分(b-2)”。):成分(b-2)中的铝原子与成分(a)中的过渡金属原子(M)的摩尔比[(b-2)/M]通常为10~5,000,优选为20~2,000。
可以以如下这样的量使用离子性化合物(b-3)(以下,也称为“成分(b-3)”。):成分(b-3)与成分(a)中的过渡金属原子(M)的摩尔比[(b-3)/M]通常为1~10,000,优选为1~5,000。
聚合温度通常为-50℃~300℃,优选为100℃~250℃,进一步优选为130℃~200℃。在上述范围的聚合温度区域中,随着温度升高,聚合时的溶液粘度降低,也容易除去聚合热。聚合压力通常为常压~10MPa表压(MPa-G),优选为常压~8MPa-G。
聚合反应可利用分批式、半连续式、连续式中的任意方法进行。此外,也可利用反应条件不同的两个以上聚合器连续地进行聚合。
得到的共聚物的分子量可通过改变聚合体系中的氢浓度、聚合温度而进行调节。此外,也可通过使用的成分(b)的量而进行调节。在添加氢时,相对于每1kg生成的共聚物,其量为0.001~5,000NL左右是适当的。
另外,共聚物(B)的分子量分布(Mw/Mn)根据使用的催化剂的结构的不同而不同。在上述式[I]这样的交联茂金属化合物的情况下,可以通过适当变更R1~R14的取代基而调节上述分子量分布。此外,也可通过利用减压蒸馏这样的现有已知的方法除去得到的聚合物的低分子量成分而调节分子量分布。
通过调节共聚物(B)的分子量和分子量分布,从而可以调节共聚物(B)的峰顶分子量和具有峰顶分子量以上的高分子量的成分中的分子量为20,000以上的成分的重量分数(即,上述“分子量为20,000以上的成分”的重量相对于上述“具有峰顶分子量以上的高分子量的成分”的重量的比例。)。另外,也可通过将分子量或分子量分布不同的多种共聚物组合而调节该重量分数。
液相聚合法中使用的聚合溶剂通常为非活性烃溶剂,优选常压下的沸点为50℃~200℃的饱和烃。作为聚合溶剂,具体而言,可举出丙烷、丁烷、戊烷、己烷、庚烷、辛烷、癸烷、十二烷、煤油等脂肪族烃;环戊烷、环己烷、甲基环戊烷等脂环族烃,可特别优选举出己烷、庚烷、辛烷、癸烷、环己烷。也可将作为聚合对象的α-烯烃自身作为聚合溶剂使用。需要说明的是,苯、甲苯、二甲苯等芳香族烃类、氯化乙烯(ethylene chloride)、氯苯、二氯甲烷等卤化烃也可作为聚合溶剂使用,但从减轻对环境的负担的观点及对人体健康的影响最小化的观点考虑,使用上述物质是不理想的。
烯烃聚合物在100℃时的运动粘度依赖于聚合物的分子量。即,如果是高分子量,则成为高粘度,如果是低分子量,则成为低粘度,因此,可通过上述的分子量调节来调节100℃时的运动粘度。进而,针对得到的聚合物,可利用现有已知的方法进行加氢(以下也称为氢化。)。通过氢化而使得到的聚合物的双键减少时,氧化稳定性及耐热性提高。
另外,以将来源于乙烯的结构单元和来源于α-烯烃的结构单元的合计作为100摩尔%时的乙烯摩尔含有率成为30~70摩尔%的范围的方式制造共聚物(B)时,进行共聚的乙烯及碳原子数为3~20的α-烯烃的装入摩尔比通常为乙烯∶α-烯烃=10∶90~99.9∶0.1,优选为乙烯∶α-烯烃=30∶70~99.9∶0.1,进一步优选为乙烯∶α-烯烃=50∶50~99.9∶0.1。
得到的乙烯-α-烯烃共聚物(B)可单独使用1种,另外,也可将2种以上的分子量或分子量分布不同的乙烯-α-烯烃共聚物(B)、单体组成不同的乙烯-α-烯烃共聚物(B)进行组合。
另外,可对乙烯-α-烯烃共聚物(B)的官能团进行接枝改性,另外,可对它们进一步进行2次改性。例如,可举出日本特开昭61-126120号公报、日本专利第2593264号公报等中记载的方法等,作为2次改性,可举出日本特表2008-508402号公报等中记载的方法等。
<润滑油组合物>
本发明的润滑油组合物含有上述润滑油基础油(A)及上述乙烯-α-烯烃共聚物(B)。
本发明的润滑油组合物在100℃时的运动粘度为20mm2/s以下。润滑油组合物在100℃时的运动粘度大于20mm2/s时,润滑油自身的油膜保持性能提高,因此,无法充分发挥通过本发明而得到的效果,另外,节省燃耗性能差。100℃时的运动粘度更优选为16mm2/s以下,进一步优选为10mm2/s以下。尤其是,为7.5mm2/s以下时,可获得高的节省燃耗性能和极其优异的剪切稳定性。该运动粘度的值是利用JIS K2283中记载的方法进行测定时得到的值。
另外,对于本发明的润滑油组合物而言,利用凝胶渗透色谱法(GPC)按照后述的方法测得的、按照标准聚苯乙烯换算的分子量中,在3,000~10,000的分子量范围内具有峰顶,具有形成该峰顶的分子量以上的高分子量的成分中的分子量为20,000以上的成分的重量分数(即,上述“分子量为20,000以上的成分”的重量相对于上述“具有形成峰顶的分子量以上的高分子量的成分”的重量的比例。以下,也简称为“分子量为20,000以上的成分的重量分数”。)为1~10%。所述3,000~10,000的分子量范围内的峰的主成分为乙烯-α-烯烃共聚物(B)。通过调节乙烯-α-烯烃共聚物(B)的分子量为20,000以上的成分的重量分数,可调节润滑油组合物中的上述重量分数。
需要说明的是,“润滑油组合物(或某种成分)在特定的分子量的范围内具有峰顶”是指,在对润滑油组合物(或某种成分)进行测定而得到的分子量分布曲线中的该范围内,存在dw/dLog(M)(M为分子量,w为具有对应的分子量的成分的重量分数。)的极大值。形成该极大值的分子量(以下,也称为“形成峰顶的分子量”。)不一定与峰顶分子量(即,形成分子量分布曲线整体中的dw/dLog(M)的最高的极大值的分子量)一致。
对于本发明的润滑油组合物而言,分子量为20,000以上的成分的重量分数大于10%时,剪切稳定性急剧显著恶化。上述重量分数优选为6%以下,进一步优选为5%以下,上述重量分数在该范围内时,可得到极其优异的剪切稳定性。
另一方面,分子量为20,000以上的成分的重量分数低于1%时,将无法获得充分的低温粘度特性。从温度粘度特性的观点考虑,分子量为20,000以上的成分的重量分数优选为2%以上,进一步优选为2.5%以上。
本发明的润滑油组合物中,上述润滑油基础油(A)与上述乙烯-α-烯烃共聚物(B)的配合比例满足目标用途的要求特性即可,没有特别限制,本发明的润滑油组合物通常以重量比((A)/(B))为99/1~50/50的比例包含上述润滑油基础油(A)和上述乙烯-α-烯烃共聚物(B)。
另外,本发明的润滑油组合物可包含极压添加剂、清净分散剂、粘度指数改进剂、抗氧化剂、防蚀剂、抗磨剂、摩擦调节剂、倾点下降剂、防锈剂及消泡剂等添加剂。
作为本发明的润滑油组合物中使用的添加剂,可例举下述物质,它们可单独使用1种或组合2种以上而使用。
极压添加剂为在各种内燃机、工业机械被置于高负荷状态下时,具有防咬损(seizing)的效果的物质的总称,没有特别限制,可例举硫醚类、亚砜类、砜类、硫代次膦酸酯(thiophosphinate)类、硫代碳酸酯类、硫化油脂、硫化烯烃等硫系极压添加剂;磷酸酯、亚磷酸酯、磷酸酯胺盐、亚磷酸酯胺类等磷酸类;氯化烃等卤素系化合物等。另外,可并用2种以上的上述化合物。
需要说明的是,在达到极压润滑条件之前,烃、或构成润滑油组合物的其他有机成分可能在加热、剪切的作用下,在极压润滑条件以前发生碳化,在金属表面形成碳化物被膜。因此,单独使用极压添加剂时,极压添加剂与金属表面的接触被碳化物被膜阻碍,可能无法期待极压添加剂的充分的效果。
对于极压添加剂而言,虽然可单独添加,但由于本发明中的润滑油组合物以共聚物之类的饱和烃为主成分,因此,从分散性的观点考虑,优选以预先将其与使用的其他添加剂一同溶解到矿物油或合成烃油等润滑油基础油中的状态进行添加的方法。具体而言,更优选下述方法:选择所谓的极压添加剂复合剂(package),添加到润滑油组合物中,所述极压添加剂复合剂是预先将极压添加剂成分等各成分配合,进而溶解到矿物油或合成烃油等润滑油基础油中而得到的。
作为优选的极压添加剂(复合剂),可举出LUBRIZOL公司制Anglamol-98A、LUBRIZOL公司制Anglamol-6043、AFTON CHEMICAL公司制HITEC1532、AFTON CHEMICAL公司制HITEC307、AFTON CHEMICAL公司制HITEC3339、RHEIN CHEMIE公司制Additin RC 9410等。
根据需要,可以以相对于润滑油组合物100质量%为0~10质量%的范围使用极压添加剂。
作为清净分散剂,可例举金属磺酸盐、金属酚盐、金属膦酸盐、琥珀酰亚胺等。根据需要,可以以相对于润滑油组合物100质量%为0~15质量%的范围使用清净分散剂。
清净分散剂也可以以与所述的其他添加剂配合并溶解到矿物油或合成烃油等润滑油中而成的DI复合剂的形式在工业上获得,例如可举出AFTON CHEMICAL公司制HITEC3419D、AFTON CHEMICAL公司制HITEC2426等。
作为抗磨剂,可例举二硫化钼等无机或有机钼化合物、石墨、硫化锑、聚四氟乙烯等。根据需要,可以以相对于润滑油组合物100质量%为0~3质量%的范围使用抗磨剂。
作为抗氧化剂,可举出2,6-二叔丁基-4-甲基苯酚等酚系、胺系的化合物。根据需要,可以以相对于润滑油组合物100质量%为0~3质量%的范围使用抗氧化剂。
作为防锈剂,可举出各种胺化合物、羧酸金属盐、多元醇酯、磷化合物、磺酸盐等化合物。根据需要,可以以相对于润滑油组合物100质量%为0~3质量%的范围使用防锈剂。
作为消泡剂,可例举二甲基硅氧烷、硅胶分散体等聚硅氧烷系化合物;醇系或酯系的化合物等。根据需要,可以以相对于润滑油组合物100质量%为0~0.2质量%的范围使用消泡剂。
作为倾点下降剂,可使用各种已知的倾点下降剂。具体而言,可使用含有有机酸酯基的高分子化合物,可特别合适地使用含有有机酸酯基的乙烯基聚合物。作为含有有机酸酯基的乙烯基聚合物,例如,可举出甲基丙烯酸烷基酯的(共)聚合物、丙烯酸烷基酯的(共)聚合物、富马酸烷基酯的(共)聚合物、马来酸烷基酯的(共)聚合物、烷基化萘等。
这样的倾点下降剂的熔点为-13℃以下,优选为-15℃,进一步优选为-17℃以下。倾点下降剂的熔点可使用差示扫描型量热计(DSC)测定。具体而言,将约5mg试样装填到铝盘中,升温至200℃,于200℃保持5分钟,然后以10℃/分钟冷却至-40℃,于-40℃保持5分钟,然后以10℃/分钟进行升温,由此时的吸热曲线求出。
此外,对于上述倾点下降剂而言,通过凝胶渗透色谱法得到的按照标准聚苯乙烯换算的重均分子量在20,000~400,000的范围内,优选在30,000~300,000的范围内,更优选在40,000~200,000的范围内。
通常可以以相对于润滑油组合物100质量%为0~2质量%的范围使用倾点下降剂。
除了上述的添加剂以外,还可根据需要而使用破乳剂、着色剂、油性剂(油性改进剂)等。
<用途>
本发明的润滑油组合物可作为工业用润滑油(齿轮油、液压油)及润滑脂用基础油使用,作为汽车用润滑油是合适的。另外,也可合适地用于差动齿轮油之类的汽车用齿轮油、或手动变速箱油、自动变速箱油、无级变速箱油、双离合变速箱油等之类的汽车用驱动油。此外,也可用于汽车发动机油、船舶气缸油。对于本发明的润滑油组合物而言,尤其是作为汽车用低粘度变速箱油,可将100℃时的运动粘度调节为7.5mm2/s以下。将该运动粘度进一步调节为6.5mm2/s以下、更优选5.5mm2/s以下时,也可发挥优异的节省燃耗性能。
实施例
以下,基于实施例进一步具体地说明本发明,但本发明不受这些实施例的限制。
[评价方法]
下述实施例及比较例等中,乙烯-α-烯烃共聚物及润滑油组合物的物性等利用以下的方法测定。
<乙烯含量(mol%)>
使用日本分光公司制傅里叶变换红外分光光度计FT/IR-610或FT/IR-6100,算出基于长链亚甲基的摇摆振动的721cm-1附近的吸收与基于丙烯的骨架振动的1155cm-1附近的吸收的吸光度比(D1155cm-1/D721cm-1),利用预先制成的标准曲线(使用ASTM D3900中的标准试样制成)求出乙烯含量(重量%)。接下来,使用得到的乙烯含量(重量%),利用下式求出乙烯含量(mol%)。
[数学式3]
<B值>
将邻二氯苯/苯-d6(4/1[vol/vol%])作为测定溶剂,在测定温度为120℃、谱宽为250ppm、脉冲重复时间为5.5秒、脉冲宽度为4.7μsec(45°脉冲)的测定条件下(100MHz,日本电子ECX400P),或在测定温度为120℃、谱宽为250ppm、脉冲重复时间为5.5秒、脉冲宽度为5.0μsec(45°脉冲)的测定条件下(125MHz,Bruker BioSpin K.K.AVANCEIIIcryo-500),测定13C-NMR波谱,基于下述式[1]算出。
[数学式4]
式[1]中,PE表示乙烯成分所占的摩尔分数,PO表示α-烯烃成分所占的摩尔分数,POE表示全部二单元组链的乙烯-α-烯烃链的摩尔分数。
<GPC测定>
对于GPC测定而言,使用Tosoh株式会社HLC-8320GPC,按照以下方式进行测定。作为分离柱,使用TSKgel SuperMultiporeHZ-M(4根),使柱温为40℃,使用四氢呋喃(和光纯药公司制)作为流动相,使展开速度为0.35ml/分钟,使试样浓度为5.5g/L,使进样量为20微升,使用差示折射计作为检测器。作为标准聚苯乙烯,使用了Tosoh公司制(PStQuick MP-M)的产品。依照通用校正的步骤,由按照标准聚苯乙烯分子量换算得到的分子量分布曲线(也称为GPC图谱)算出乙烯-α-烯烃共聚物的峰顶分子量、及润滑油组合物的在3,000~10,000的分子量范围内形成峰顶的分子量。
另外,关于(B)乙烯-α-烯烃共聚物、聚-α-烯烃、及润滑油组合物中的分子量为20,000以上的成分的重量分数,对在得到的GPC图谱与基线之间形成的区域进行划分,由此,基于经划分的区域的面积,算出检测到的下述成分中的分子量为20,000以上的成分的重量分数,所述成分具有分子量3,000~10,000中的形成峰顶的分子量以上的高分子量。
<分子链双键量>
将邻二氯苯-d4作为测定溶剂,在测定温度为120℃、谱宽为20ppm、脉冲重复时间为7.0秒、脉冲宽度为6.15μsec(45°脉冲)的测定条件下,测定1H-NMR波谱(400MHz,日本电子ECX400P),使用溶剂峰(邻二氯苯7.1ppm)作为化学位移基准,由在0~3ppm处观测到的主峰与在4~6ppm处观测到的来源于双键的峰的积分值的比率算出每1000个碳原子的双键量(本说明书中,称为“分子链双键量”。)(个/1000C)。
<熔点>
使用Seiko Instruments Inc.的X-DSC-7000,向可简易密封的铝样品盘中放入约8mg的乙烯-α-烯烃共聚物,配置到DSC样品槽中,在氮气氛下,以10℃/分钟将DSC样品槽从室温升温至150℃,接下来,于150℃保持5分钟,然后以10℃/分钟进行降温,将DSC样品槽冷却至-100℃(降温过程)。接下来,于-100℃保持5分钟,然后以10℃/分钟进行升温,由在两个过程中得到的焓曲线确认有无吸热或放热峰。在观测不到峰或熔化热(ΔH)的值为1J/g以下时,视为未观测到熔点(Tm)。熔点(Tm)、及熔化热(ΔH)的求解方法基于JIS K7121。
<含氯量>
使用Thermo Fisher Scientific公司的ICS-1600,在试样舟皿中放入乙烯-α-烯烃共聚物,在Ar/O2气流中,于900℃的燃烧炉设定温度进行燃烧分解。将此时产生的气体吸收于吸收液中,用离子色谱法进行定量。
<粘度特性>
100℃运动粘度及粘度指数利用JIS K2283中记载的方法进行测定、计算。
<剪切试验>
对于润滑油组合物的剪切稳定性而言,按照CRC L-45-T-93中记载的方法,使用KRL剪切试验机进行评价。但是,试验时间不是所记载的20小时,而是设定为100小时,在试验温度为60℃、轴承转速为1450rpm的剪切条件下,评价下式表示的剪切试验粘度降低率。
剪切试验粘度降低率(%)=(剪切前的100℃运动粘度-剪切后的100℃运动粘度)/剪切前的100℃运动粘度×100
<-40℃粘度>
作为低温粘度特性,按照ASTM D2983,于-40℃,利用布氏粘度计(Brookfield viscometer)测定-40℃粘度。
[乙烯-α-烯烃共聚物(B)的制造]
乙烯-α-烯烃共聚物(B)按照以下的聚合例制造。需要说明的是,针对得到的乙烯-α-烯烃共聚物(B),根据需要,利用下述方法实施氢化操作。
<氢化操作>
向内容积1L的不锈钢制高压釜中添加100mL的0.5质量%Pd/氧化铝催化剂的己烷溶液及500mL乙烯-α-烯烃共聚物的30质量%己烷溶液,将高压釜密闭后,进行氮置换。接下来,一边搅拌一边升温至140℃,对体系内进行氢置换,然后利用氢升压至1.5MPa,实施15分钟氢化反应。
<茂金属化合物的合成>
双(η5-1,3-二甲基环戊二烯基)二氯化锆利用日本特公平6-62642号公报中记载的方法合成。
<合成例1>[甲基苯基亚甲基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆的合成
(i)6-甲基-6-苯基富烯(fulvene)的合成
在氮气氛下,向200mL三颈瓶中添加7.3g(101.6mmol)环戊二烯锂及100mL无水四氢呋喃并进行搅拌。在冰浴中冷却溶液,滴加15.0g(111.8mmol)苯乙酮。然后,在室温下搅拌20小时,用稀盐酸水溶液将得到的溶液猝灭(quench)。添加100mL己烷,萃取可溶成分,用水、饱和食盐水洗涤该有机层,然后用无水硫酸镁干燥。然后,馏出溶剂,用柱色谱法(己烷)将得到的粘性液体分离,得到目标物(红色粘性液体)。
(ii)甲基(环戊二烯基)(2,7-二叔丁基芴基)(苯基)甲烷的合成
在氮气氛下,向100mL三颈瓶中添加2.01g(7.20mmol)2,7-二叔丁基芴及50mL无水叔丁基甲基醚。一边在冰浴中冷却一边缓缓添加4.60mL(7.59mmol)正丁基锂/己烷溶液(1.65M),在室温下搅拌16小时。添加1.66g(9.85mmol)6-甲基-6-苯基富烯,然后在加热回流下搅拌1小时。一边在冰浴中冷却一边缓缓添加50mL水,将得到的两层溶液移至200mL分液漏斗。添加50mL乙醚并振荡数次后,除去水层,用50mL水洗涤有机层3次,用50mL饱和食盐水洗涤有机层1次。用无水硫酸镁进行30分钟干燥,然后在减压下馏出溶剂。向添加少量的己烷而得到的溶液照射超声波,结果析出固体,因此,收集该固体并用少量的己烷洗涤。在减压下将其干燥,以白色固体形式得到2.83g甲基(环戊二烯基)(2,7-二叔丁基芴基)(苯基)甲烷。
(iii)[甲基苯基亚甲基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆的合成
在氮气氛下,向100mL舒伦克瓶(Schlenk flask)中依序添加1.50g(3.36mmol)甲基(环戊二烯基)(2,7-二叔丁基芴基)(苯基)甲烷、50mL无水甲苯及570μL(7.03mmol)THF。一边在冰浴中冷却一边缓缓添加4.20mL(6.93mmol)正丁基锂/己烷溶液(1.65M),于45℃搅拌5小时。在减压下馏出溶剂,添加40mL无水乙醚,形成红色溶液。一边在甲醇/干冰浴中冷却一边添加728mg(3.12mmol)四氯化锆,一边缓缓升温至室温,一边搅拌16小时,结果,得到橙红色浆料。将在减压下馏出溶剂而得到的固体拿进手套箱内,用己烷洗涤后,用二氯甲烷萃取。在减压下馏出溶剂而将其浓缩,然后添加少量的己烷,于-20℃放置,结果析出了橙红色固体。用少量的己烷洗涤该固体,然后在减压下将其干燥,由此,以橙红色固体形式得到1.20g[甲基苯基亚甲基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆。
<聚合例1>
在经充分氮置换的内容积2L的不锈钢制高压釜中装入760ml庚烷、120g丙烯,将体系内的温度升温至150℃,然后通过供给0.85MPa氢、0.19MPa乙烯而使总压力为3MPaG。接下来,用氮压入0.4mmol三异丁基铝、0.0002mmol[甲基苯基亚甲基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆、及0.002mmol的四(五氟苯基)硼酸N,N-二甲基苯胺鎓,将搅拌转速设为400rpm,由此开始进行聚合。然后,通过连续地供给乙烯从而将总压力保持为3MPaG,于150℃进行5分钟聚合。通过向体系内添加少量的乙醇从而终止聚合,然后将未反应的乙烯、丙烯、氢清除。针对得到的聚合物溶液,用1000ml的0.2mol/L的盐酸洗涤3次,接下来,用1000ml蒸馏水洗涤3次,用硫酸镁干燥,然后减压馏出溶剂。在80℃的减压下将得到的聚合物干燥10小时。接下来,通过氢化操作,得到聚合物1。
聚合物1的分子链双键量小于0.1个/1000C,氯含量小于0.1ppm。聚合物1的乙烯含量为48.5mol%,峰顶分子量为5,218,具有峰顶分子量以上的高分子量的成分中的分子量为20,000以上的成分的重量分数为1.22%,B值为1.2,100℃运动粘度为155mm2/s,未观测到熔点(熔融峰)。
<聚合例2>
在经充分氮置换的内容积2L的不锈钢制高压釜中装入750mL庚烷及125g丙烯,将体系内的温度升温至150℃,然后,通过供给0.69MPa氢、0.23MPa乙烯而使总压力为3MPaG。接下来,用氮压入0.4mmol三异丁基铝、0.0001mmol[甲基苯基亚甲基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆及0.001mmol四(五氟苯基)硼酸N,N-二甲基苯胺鎓,将搅拌转速设为400rpm,由此开始进行聚合。然后,通过仅连续地供给乙烯从而将总压力保持为3MPaG,于150℃进行5分钟聚合。通过向体系内添加少量的乙醇从而终止聚合,然后将未反应的乙烯、丙烯、氢清除。针对得到的聚合物溶液,用1000mL的0.2mol/l的盐酸洗涤3次,接下来,用1000mL蒸馏水洗涤3次,用硫酸镁干燥,然后减压馏出溶剂。在80℃的减压下将得到的聚合物干燥一夜,得到52.2g乙烯-丙烯共聚物。接下来,通过氢化操作,得到聚合物2。
聚合物2的分子链双键量小于0.1个/1000C,氯含量小于0.1ppm。聚合物2的乙烯含量为49.7mol%,峰顶分子量为6,186,具有峰顶分子量以上的高分子量的成分中的分子量为20,000以上的成分的重量分数为2.92%,B值为1.2,100℃运动粘度为281mm2/s,未观测到熔点(熔融峰)。
<聚合例3>
在经充分氮置换的内容积2L的不锈钢制高压釜中装入710ml庚烷、145g丙烯,将体系内的温度升温至150℃,然后,通过供给0.43MPa氢、0.26MPa乙烯而使总压力为3MPaG。接下来,用氮压入0.4mmol三异丁基铝、0.0001mmol[甲基苯基亚甲基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆、及0.001mmol四(五氟苯基)硼酸N,N-二甲基苯胺鎓,将搅拌转速设为400rpm,由此开始进行聚合。然后,通过连续地供给乙烯从而将总压力保持为3MPaG,于150℃进行5分钟聚合。通过向体系内添加少量的乙醇从而终止聚合,然后将未反应的乙烯、丙烯、氢清除。针对得到的聚合物溶液,用1000ml的0.2mol/L的盐酸洗涤3次,接下来,用1000ml蒸馏水洗涤3次,用硫酸镁干燥,然后减压馏出溶剂。在80℃的减压下将得到的聚合物干燥10小时。接下来,通过氢化操作,得到聚合物3。
聚合物3的分子链双键量小于0.1个/1000C,氯含量小于0.1ppm。聚合物3的乙烯含量为50.4mol%,峰顶分子量为7,015,具有峰顶分子量以上的高分子量的成分中的分子量为20,000以上的成分的重量分数为5.24%,B值为1.2,100℃运动粘度为411mm2/s,未观测到熔点(熔融峰)。
<聚合例4>
在经充分氮置换的内容积2L的不锈钢制高压釜中装入910mL庚烷及45g丙烯,将体系内的温度升温至130℃,然后通过供给2.24MPa氢、0.09MPa乙烯而使总压力为3MPaG。接下来,用氮压入0.4mmol三异丁基铝、0.0006mmol[甲基苯基亚甲基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆及0.006mmol四(五氟苯基)硼酸N,N-二甲基苯胺鎓,将搅拌转速设为400rpm,由此开始进行聚合。然后,通过仅连续地供给乙烯从而将总压力保持为3MPaG,于130℃进行5分钟聚合。通过向体系内添加少量的乙醇从而终止聚合,然后将未反应的乙烯、丙烯、氢清除。针对得到的聚合物溶液,用1000mL的0.2mol/l的盐酸洗涤3次,接下来,用1000mL蒸馏水洗涤3次,用硫酸镁干燥,然后减压馏出溶剂。在80℃的减压下将得到的聚合物干燥一夜后,进一步使用SHINKO-PANTEC CO.,LTD.制2-03型薄膜蒸馏装置,将真空度保持为400Pa,以180℃的设定温度、3.1ml/min的流量进行薄膜蒸馏,得到22.2g乙烯-丙烯共聚物。接下来,通过氢化操作,得到聚合物4。
聚合物4的分子链双键量小于0.1个/1000C,氯含量小于0.1ppm。聚合物4的乙烯含量为51.9mol%,峰顶分子量为2,572,具有峰顶分子量以上的高分子量的成分中的分子量为20,000以上的成分的重量分数为0.05%,B值为1.2,100℃运动粘度为40mm2/s,未观测到熔点(熔融峰)。
<聚合例5>
在经充分氮置换的容量2升的带有搅拌叶片的连续聚合反应器中,装入1升经脱水纯化的己烷,以500ml/h的量连续地供给已调节为96mmol/L的乙基倍半氯化铝(Al(C2H5)1.5·Cl1.5)的己烷溶液1小时,然后进一步以500ml/h的量连续地供给作为催化剂的已调节为16mmol/l的VO(OC2H5)Cl2的己烷溶液,并以500ml/h的量连续地供给己烷。另一方面,从聚合器上部连续地取出聚合液,使得聚合器内的聚合液一直保持为1升。接下来,使用鼓泡管,以35L/h的量供给乙烯气体,以35L/h的量供给丙烯气体,以80L/h的量供给氢气。通过使制冷剂在被安装于聚合器外部的外罩(jacket)中循环从而于35℃进行共聚反应。针对在上述条件下得到的包含乙烯-丙烯共聚物的聚合溶液,用100mL的0.2mol/l的盐酸洗涤3次,接下来用100mL蒸馏水洗涤3次,用硫酸镁干燥,然后减压馏出溶剂。在130℃的减压下将得到的聚合物干燥一夜。
通过以上的操作而得到的聚合物5(乙烯-丙烯共聚物)的乙烯含量为54.9mol%,峰顶分子量为4,031,具有峰顶分子量以上的高分子量的成分中的分子量为20,000以上的成分的重量分数为0.32%,B值为1.2,100℃运动粘度为102mm2/s,未观测到熔点(熔融峰)。另外,分子链双键量为0.1个/1000C,氯含量为15ppm。
<聚合例6>
在经充分氮置换的内容积2L的不锈钢制高压釜中装入710mL庚烷及145g丙烯,将体系内的温度升温至150℃,然后,通过供给0.40MPa氢、0.27MPa乙烯而使总压力为3MPaG。接下来,用氮压入0.4mmol三异丁基铝、0.0001mmol[甲基苯基亚甲基(η5-环戊二烯基)(η5-2,7-二叔丁基芴基)]二氯化锆及0.001mmol四(五氟苯基)硼酸N,N-二甲基苯胺鎓,将搅拌转速设为400rpm,由此开始进行聚合。然后,通过仅连续地供给乙烯从而将总压力保持为3MPaG,于150℃进行5分钟聚合。通过向体系内添加少量的乙醇从而终止聚合,然后将未反应的乙烯、丙烯、氢清除。针对得到的聚合物溶液,用1000mL的0.2mol/l的盐酸洗涤3次,接下来,用1000mL蒸馏水洗涤3次,用硫酸镁干燥,然后减压馏出溶剂。在80℃的减压下将得到的聚合物干燥一夜,得到52.2g烯-丙烯共聚物。接下来,通过氢化操作,得到聚合物6。
聚合物6的分子链双键量小于0.1个/1000C,氯含量小于0.1ppm。聚合物6的乙烯含量为53.1mol%,峰顶分子量为8,250,具有峰顶分子量以上的高分子量的成分中的分子量为20,000以上的成分的重量分数为12.90%,B值为1.2,100℃运动粘度为608mm2/s,未观测到熔点(熔融峰)。
<聚合例7>
在经充分氮置换的容量2升的带有搅拌叶片的连续聚合反应器中,装入1升经脱水纯化的己烷,以500ml/h的量连续地供给已调节为96mmol/L的乙基倍半氯化铝(Al(C2H5)1.5·Cl1.5)的己烷溶液1小时,然后进一步以500ml/h的量连续地供给作为催化剂的已调节为16mmol/l的VO(OC2H5)Cl2的己烷溶液,并以500ml/h的量连续地供给己烷。另一方面,从聚合器上部连续地取出聚合液,使得聚合器内的聚合液一直保持为1升。接下来,使用鼓泡管,以47L/h的量供给乙烯气体,以47L/h的量供给丙烯气体,以20L/h的量供给氢气。通过使制冷剂在被安装于聚合器外部的外罩中循环从而于35℃进行共聚反应。针对在上述条件下得到的包含乙烯-丙烯共聚物的聚合溶液,用100mL的0.2mol/l的盐酸洗涤3次,接下来用100mL蒸馏水洗涤3次,用硫酸镁干燥,然后减压馏出溶剂。在130℃的减压下将得到的聚合物干燥一夜。
通过以上的操作而得到的聚合物7(乙烯-丙烯共聚物)的乙烯含量为54.9mol%,峰顶分子量为12,564,具有峰顶分子量以上的高分子量的成分中的分子量为20,000以上的成分的重量分数为44.15%,B值为1.2,100℃运动粘度为2,040mm2/s,未观测到熔点(熔融峰)。另外,分子链双键量为0.1个/1000C,氯含量为8ppm。
<聚合例8>
在经充分氮置换的内容积2L的不锈钢制高压釜中装入190ml庚烷、405g丙烯,将体系内的温度升温至80℃,然后通过供给100Nml氢、0.20MPa乙烯而使总压力为3MPaG。接下来,用氮压入0.4mmol三异丁基铝、0.0003mmol双(η5-1,3-二甲基环戊二烯基)二氯化锆、及0.003mmol四(五氟苯基)硼酸N,N-二甲基苯胺鎓,将搅拌转速设为400rpm,由此开始进行聚合。然后,通过连续地供给乙烯从而将总压力保持为3MPaG,于80℃进行5分钟聚合。通过向体系内添加少量的乙醇从而终止聚合,然后将未反应的乙烯、丙烯、氢清除。针对得到的聚合物溶液,用1000ml的0.2mol/L的盐酸洗涤3次,接下来,用1000ml蒸馏水洗涤3次,用硫酸镁干燥,然后减压馏出溶剂。在80℃的减压下将得到的聚合物干燥10小时。接下来,通过氢化操作,得到聚合物8。
聚合物8的分子链双键量小于0.1个/1000C,氯含量小于0.1ppm。聚合物8的乙烯含量为52.2mol%,峰顶分子量为6,401,具有峰顶分子量以上的高分子量的成分中的分子量为20,000以上的成分的重量分数为12.97%,B值为1.2,100℃运动粘度为408mm2/s,未观测到熔点(熔融峰)。
[润滑油组合物的制备]
以下的润滑油组合物的制备中使用的乙烯-α-烯烃共聚物(B)以外的成分如下所述。
润滑油基础油:
100℃运动粘度为5.8mm2/s的合成烃油PAO(NESTE公司制NEXBASE2006,PAO-6),
100℃运动粘度为3.0mm2/s的API(American Petroleum Institute)第二类(Group II)矿物油(NESTE公司制NEXBASE3030,矿物油-A),以及
作为脂肪酸酯的大八化学公司制己二酸二异癸酯(DIDA)。
极压添加剂复合剂:LUBRIZOL公司制ANGLAMOL-98A(EP)。
倾点下降剂:BASF公司制IRGAFLO 720P(PPD)。
作为聚-α-烯烃,使用以下的物质。
PAO-100:以碳原子数为6以上的α-烯烃为单体,100℃运动粘度为100mm2/s、峰顶分子量为4,325、峰顶以上的高分子量成分中的分子量为20,000以上的重量分数为0.20%的、使用酸催化剂得到的PAO(ExxonMobil Chemical公司制Spectrasyn 100)。
mPAO-100:以1-癸烯为单体,100℃运动粘度为100mm2/s、峰顶分子量为5,202、峰顶以上的高分子量成分中的分子量为20,000以上的重量分数为0.22%的、使用茂金属系催化剂得到的PAO(Ineos Oligmers公司制Durasyn 180R)。
mPAO-300:以1-辛烯为单体,100℃运动粘度为302mm2/s、峰顶分子量为7,229、峰顶以上的高分子量成分中的分子量为20,000以上的重量分数为5.45%的、使用茂金属系催化剂得到的PAO。按照WO2011/142345号小册子的聚合例1中记载的方法得到该聚合物。未观测到熔点(熔融峰)。
<汽车用齿轮油>
实施例1~3中,按照汽车工程师协会(Society of Automobile Engineers,SAE)的齿轮油粘度标准90,进行配合调整,以使得100℃运动粘度约为14mm2/s。将下述实施例及比较例中得到的润滑油组合物的配合及润滑油特性示于表2。该粘度标准为可合适地用于汽车用差动齿轮油、以及卡车、公共汽车用手动变速箱油等的粘度标准。
[实施例1]
以使作为乙烯-α-烯烃共聚物(B)的聚合例1中得到的共聚物为28.0质量%、作为润滑油基础油(A)的DIDA为15.0质量%、极压添加剂复合剂(EP)为6.5质量%的方式进行配合,进而向其中添加作为润滑油基础油(A)的PAO-6,使得润滑油组合物的整体为100质量%,制备润滑油组合物。
[实施例2]
将聚合物1替换为18.4质量%的聚合物2,除此之外,与实施例1同样地制备润滑油组合物。
[实施例3]
将聚合物1替换为17.0质量%的聚合物3,除此之外,与实施例1同样地制备润滑油组合物。
[比较例1]
将聚合物1替换为44.7质量%的聚合物4,除此之外,与实施例1同样地制备润滑油组合物。测定得到的润滑油组合物的分子量,结果,在GPC图谱中,在3,000~10,000的分子量范围内不存在峰。需要说明的是,观察到分子量2,670的极大值(推断其来源于聚合物4),将具有分子量2,670以上的高分子量的成分中的分子量为20,000以上的成分的重量分数0.06%示于表2的“分子量为20,000以上的成分的重量分数”的栏中。
[比较例2]
将聚合物1替换为29.8质量%的聚合物5,除此之外,与实施例1同样地制备润滑油组合物。
[比较例3]
将聚合物1替换为14.2质量%的聚合物6,除此之外,与实施例1同样地制备润滑油组合物。
[比较例4]
将聚合物1替换为10.7质量%的聚合物7,除此之外,与实施例1同样地制备润滑油组合物。测定得到的润滑油组合物的分子量,结果,在3,000~10,000的分子量范围内不存在峰,在分子量13,030处具有推断是基于聚合物7的极大值。将具有分子量13,030以上的高分子量的成分中的分子量为20,000以上的成分的重量分数44.07%示于表2的“分子量为20,000以上的成分的重量分数”的栏中。
[比较例5]
将聚合物1替换为17.2质量%的聚合物8,除此之外,与实施例1同样地制备润滑油组合物。
[比较例6]
代替作为乙烯-α-烯烃共聚物(B)的聚合物1,配合30.7质量%的PAO-100,除此之外,与实施例1同样地制备润滑油组合物。
[比较例7]
代替作为乙烯-α-烯烃共聚物(B)的聚合物1,配合35.6质量%的mPAO-100,除此之外,与实施例1同样地制备润滑油组合物。
[比较例8]
代替作为乙烯-α-烯烃共聚物(B)的聚合物(1),配合24.7质量%的mPAO-300,除此之外,与实施例1同样地制备润滑油组合物。
对于实施例1~3而言,-40℃时的布氏(Brookfield)粘度均低于40,000mPa·s,与比较例1、比较例2相比,低温粘度特性优异,所述比较例1中,乙烯-α-烯烃共聚物的峰顶分子量低于3,000,所述比较例2中,虽然乙烯-α-烯烃共聚物的峰顶分子量在3,000~10,000的范围内,但润滑油组合物中的分子量为20,000以上的成分的重量分数低于1%。
另外,对于实施例1~3而言,试验时间为100小时的剪切试验粘度降低率均低于3%,与比较例4、以及比较例3及比较例5相比,剪切稳定性非常优异,所述比较例4中,乙烯-α-烯烃共聚物的峰顶分子量大于10,000,所述比较例3及比较例5中,虽然乙烯-α-烯烃共聚物的峰顶分子量在3,000~10,000的范围内,但润滑油组合物中的分子量为20,000以上的成分的重量分数大于10%。尤其是,对实施例3与比较例5进行比较可知,虽然乙烯-α-烯烃共聚物的100℃运动粘度大致相等,但由于分子量为20,000以上的成分的重量分数不同,因而剪切稳定性差异很大。
此外,使用聚-α-烯烃代替乙烯-α-烯烃共聚物(B)时,α-烯烃侧链受到剪切应力的很大影响,剪切稳定性明显差。
由图1及图2所示的实施例2和比较例3中的润滑油组合物的剪切试验前(实线)与剪切试验后(虚线或点线)的GPC图谱的比较可知,通过剪切试验,分子量为20,000以上的成分选择性地受到剪切应力,发生分子切断。
对于比较例3~7的润滑油组合物而言,在剪切试验后均不满足齿轮油粘度标准SAE 90,为了在剪切试验后满足该标准,必须在配合制备时相应地增加粘度以弥补各粘度降低率,该粘度的增加导致低温粘度特性恶化。可知不需要进行该粘度增加的本发明的润滑油组合物在节省燃耗方面非常优异。
<汽车用低粘度变速箱油>
在实施例4~6中,进行配合制备以使得100℃运动粘度约为6mm2/s。将下述实施例及比较例中得到的润滑油组合物的润滑油特性示于表3。该配合为可合适地用于汽车用手动变速箱油、自动变速箱油、无级变速箱油、以及双离合变速箱油等的粘度范围内的配合。
[实施例4]
添加13.5质量%作为乙烯-α-烯烃共聚物(B)的聚合物1、0.5质量%倾点下降剂(PPD),向其中添加作为润滑油基础油(A)的矿物油-A,以使得润滑油组合物整体成为100质量%,制备润滑油组合物。
[实施例5]
将聚合物1替换为11.6质量%的聚合物2,除此之外,与实施例4同样地配合制备润滑油组合物。
[实施例6]
将聚合物1替换为10.4质量%的聚合物3,除此之外,与实施例4同样地配合制备润滑油组合物。
[比较例9]
将聚合物1替换为16.1质量%的聚合物5,除此之外,与实施例4同样地配合制备润滑油组合物。
[比较例10]
将聚合物1替换为9.3质量%的聚合物6,除此之外,与实施例4同样地配合制备润滑油组合物。
[比较例11]
代替作为乙烯-α-烯烃共聚物(B)的聚合物1,配合18.4质量%的PAO-100,除此之外,与实施例4同样地配合制备润滑油组合物。
[比较例12]
代替作为乙烯-α-烯烃共聚物(B)的聚合物1,配合21.4质量%的mPAO-100,除此之外,与实施例4同样地配合制备润滑油组合物。
对于实施例4~6而言,-40℃时的布氏粘度均低于10,000mPa·s,与比较例9相比,低温粘度特性优异,所述比较例9中,虽然乙烯-α-烯烃共聚物(B)的峰顶分子量在3,000~10,000的范围内,但润滑油组合物中的分子量为20,000以上的成分的重量分数小于1%。
另外,对于100℃运动粘度为7.5mm2/s以下的润滑油组合物而言,实施例4~6中,试验时间为100小时的剪切试验粘度降低率均小于1%,与比较例10相比,剪切稳定性非常优异,所述比较例10中,虽然乙烯-α-烯烃共聚物(B)的峰顶分子量在3,000~10,000的范围内,但润滑油组合物中的分子量为20,000以上的成分的重量分数大于10%。即,通过本发明,可实现在剪切应力下几乎不发生粘度降低的润滑油。
此外,使用聚-α-烯烃代替乙烯-α-烯烃共聚物(B)时,α-烯烃侧链受到剪切应力的很大影响,剪切稳定性明显差。
另外,本发明的润滑油组合物与现有技术的润滑油相比,可进一步降低制造时的粘度(初始粘度),因此,从节省燃耗的观点考虑也是优异的。
另外,即使通过将实施例1中使用的极压添加剂复合剂替换为各种添加剂、例如不含分子量为20,000以上的成分的自动变速箱油用添加剂复合剂、无级变速箱油用添加剂复合剂从而将本发明的润滑油组合物用作自动变速箱油、无级变速箱油,也可获得与实施例1的润滑油组合物同样的效果。
- 还没有人留言评论。精彩留言会获得点赞!