一种合成钽基氮(氧)化物纳米颗粒的方法及其纳米颗粒与流程
本发明属于纳米材料技术领域,特别涉及一种可控合成钽基氮(氧)化物纳米颗粒的方法及其纳米颗粒。
背景技术:
以氮化钽为代表的过渡金属氮化物由于其独特的类贵金属的电子结构和催化性能、良好的热稳定性,成为近一、二十年来最有应用前景的催化材料之一。特别是近年来在伴随着石油价格快速上涨、能源和资源的高效和绿色化利用的呼声日益高涨,对氮化物及氮氧化物的光催化性能和涉氢催化反应的开发进一步引起了人们的关注。除此而外,由于此类过渡金属氮化物、氮氧化物合成原料较为便宜易得,在节约贵金属宝贵资源,降低催化反应成本也具有重要意义。金属氮化物催化剂在包括肼分解、NH3合成和分解、加氢精制、烃类异构、选择加氢、CO2和CO加氢、F-T合成等方面得到广泛的应用,在能源和化学工业催化反应中展现了巨大潜力(Volpe L,Boudart M.Ammonia synthesis on molybdenum nitride.J.Phys.Chem.1986,9:4874-4877;J.S.J.Hargreaves,Coordination Chemistry Reviews.2013,257,2015-2031;X.J.Lang,X.D.Chen,J.C.Zhao,Chemical Society Reviews.2014,43,473-486;A.Salamat,A.L.Hector,P.Kroll,P.F.McMillan,Coordination Chemistry Reviews.2013,257,2063-2072;N.E.Breese,Structure and Bonding,Crystal Chemistry of Inorganic Nitride,Springer,Berlin,1992;H.Morkoc,S.N.Mohammad,Science.1995,267,51;V.Heine,Phys.Rev.A 1967,153,673;E.Furimsky,Applied Catalysis a-General.2003,240,1-28;A.M.Alexander,J.S.J.Hargreaves,Chem.Soc.Rev.2010,39,4388-4401;Q.S.Gao,N.Liu,S.N.Wang,Y.Tang,Nanoscale.2014,6,14106;F.Monnet,Y.Schuurman,F.C.S.Aires,J.C.Bertolini,C.Mirodatos,Catal.Today.2001,64, 51-58;M.M.O.Thotiyl,T.R.Kumar,S.Sampath,J.Phys.Chem.C.2010,114,17934-17941;V.Molinari,C.Giordano,M.Antonietti,D.Esposito,J.Am.Chm.Soc.2014,136,1758-1761;F.Cardenas-Lizana,D.Lamey,N.Perret,S.Gomez-Quero,L.Kiwi-Minsker,M.A.Keane,Catalysis Communications.2012,21,46-51;K.Maeda,K.Domen,Journal of Physical Chemistry C.2007,111,7851-7861;R.Asahi,T.Morikawa,T.Ohwaki,K.Aoki,Y.Taga,Science.2001,293,269-271;Q.S.Gao,C.Giordano,M.Antonietti,Small.2011,7,3334-3340;Y.G.Su,J.Y.Lang,L.P.Li,K.Guan,C.F.Du,L.M.Peng,D.Han,X.J.Wang,J.Am.Chem.Soc.2013,135,11433-11436;Q.S.Gao,S.N.Wang,Y.C.Ma,Y.Tang,C.Giordano,M.Antonietti,Angew.Chem.Int.Ed.2012,51,961-965;R.Abe,M.Higashi,K.Domen,J.Am.Chem.Soc.2010,132,11828-11829;Q.S.Gao,C.Giordano,M.Antonietti,Angew.Chem.Int.Ed.2012,51,11740-11744.)。例如,近几年来,国内外众多学者已在大力研究金属氮化物以及其催化性能,发现单金属氮化物、双金属氮化物以及负载型金属氮化物都是优良的加氢和脱氢催化剂,在许多涉氢反应中:F-T合成、CO氧化、CO加氢、NO还原等反应中都表现出了良好的催化能力。另一方面,由于钽基氮氧化物及氮化物具有半导体性质,可以吸收太阳光谱中的可见光,其能带结构也能进行控制,这使得它在光催化分解水反应和光降解有机污染物及无机染料工业方面有广泛应用。
从文献报道看,传统的金属氮化物合成方法大约可以分为以下几类:1)程序升温法,由设定的程序来升温氮化前驱体,但是由于温度高(>750℃),合成条件苛刻,难以实现产业化。2)热分解法,利用金属有机化合物或金属有机化合物的盐在载气下加热分解合成金属氮化物,但是对有些金属难合成金属有机化合物及其盐,有局限性。3)固态反应法,以金属粉末、金属氧化物、或金属盐做金属源,NaN3等为氮源,固态反应后合成金属氮化物,但是此法需要的压力较高,且所使用氮源价格昂贵。4)球磨法,是在一定的气氛下通过球磨使原料充分混合并细化、反应,但是由于反应时间长,原料难以完全反应使其难大规模使用。此外还有电化学法、化学气相沉积法、超声笔合成法、溶胶凝胶法等超声波分散等报道,但同样存在产量小,反应不充分,产物颗粒 较大等难题(E.Orhan,F.Tessier,R.Marchand,Solid State Sciences.2002,4,1071-1076;M.Kerlau,O.Merdrignac-Conanec,M.Guilloux-Viry,A.Perrin,Solid State Sciences.2004,6,101-107;J.B.Claridge,A.P.E.York,A.J.Brungs,M.L.H.Green,Chem.Mater.2000,12,132-142;H.Zhao,M.Lei,X.Chen,W.Tang,J.Mater.Chem.2006,16,4407-4412;J.Buha,I.Djerdj,M.Antonietti,M.Niederberger,Chem.Mater.2007,19,3499-3505;Kasahara A,Nukumizu K,Hitoki G,Takata T,Kondo J N,Hara M,Kobayashi H,Domen K.Photoreaetions on LaTIO2N under visible light irradiation.Journal of Physical Chemistry.A.2002,106:6750-6752;Tessier F,Marehand R.Ternary and higher order rare-earth nitride materials:synthesis and characterization of ionic-covalent oxynitride powders.J.Solid State Chem.2003,171:143-151;Volpe L,Boudart M.Compounds of molybdenum and tungsten with high specific surface area:I.nitrides.J.Solid State Chem.1985,59:332-347;Wise R S,Markel E J.Catalytic NH3decomposition by topotactic molybdenum oxides and nitrides:Effect on temperature programmed γ-Mo2N synthesis.J.Catal.1994,145:335-343.)。
因此,利用新的合成方法高效、精确制备的钽基氮氧化物及氮化物新型纳米催化剂,是目前该领域的最有兴趣的研究课题之一。可控氮化合成的钽基氮氧化物及氮化物不仅反应条件温和环保,并且克服了过度氮化的问题,从而能够控制能带结构,其次外观非常漂亮,呈现五彩缤纷的颜色,过渡金属氮氧化物这些有趣的性质决定了其在光催化领域和染料工业具有开发潜力和应用前景。然而,目前在有效实现可控氮化方面尚无报道。
技术实现要素:
为了克服上述现有技术的缺点与不足,本发明的首要目的在于提供一种可控合成钽基氮(氧)化物纳米颗粒的方法。
本发明的另一目的在于提供上述方法制备得到的钽基氮(氧)化物纳米颗粒。所得纳米颗粒的粒径均在10nm左右。
本发明再一目的在于提供上述钽基氮(氧)化物纳米颗粒在精细化工催化 反应、燃料电池电极催化材料、传感器以及电学材料等领域中的应用。
本发明的目的通过下述方案实现:
一种可控合成钽基氮(氧)化物纳米颗粒的方法,包括以下步骤:
将钽盐溶解于有机醇中,加入碱土金属盐和有机氮源,搅拌得到前驱体,置于惰性气氛中高温焙烧后,盐酸溶液处理,得到钽基氮(氧)化物纳米颗粒。
所述碱土金属盐和钽盐的用量以碱土金属离子与钽原子的摩尔比为准,优选为(0.1~10):1,更优选为(0.1~5):1。
所述有机氮源和钽盐的用量以氮元素与钽原子的摩尔比为准,优选为(1.0~10):1,更优选为(2~7):1。
所述的钽盐可为五氯化钽、五溴化钽、氧化钽和五乙氧基钽等中的至少一种,优选为五氯化钽。
所述的碱土金属盐可为氯化锶、碳酸锶、硝酸锶、氯化钡、碳酸钡、硝酸钡、氯化镁、硫酸镁和硝酸镁等中的至少一种,优选为氯化锶。
所述的有机氮源可为尿素、单氰胺、二氰二胺和三聚氰胺等中的至少一种,优选为尿素。所述有机氮源优选在碱土金属盐溶解后再加入。
所述高温焙烧优选指在650~850℃下焙烧2~10h,更优选在700℃下焙烧5h,升温速率优选为1~10℃/min。
所述盐酸溶液处理指高温焙烧后的产物置于盐酸溶液中搅拌24~48h,优选为48h。所述利用盐酸溶液处理是为了除去多余的碱土金属离子,因此所用盐酸溶液浓度可任意调节,优选为1mol/L。所述盐酸溶液处理后优选再用无水乙醇洗涤多次,烘干,得到纯化后产物。
所述的有机醇用于提供溶液反应环境,为本领域常用醇类溶剂即可,如甲醇、乙醇、异丙醇等。
所述搅拌得到前驱体,优选在室温下搅拌4~48h,至形成凝胶状体系,得到前驱体。
所述的惰性气氛可以是氩气、氮气或氦气。
上述方法制备得到的钽基氮(氧)化物纳米颗粒,该颗粒粒径均为10nm左右。
本发明方法制备得到的钽基氮氧化物及钽基氮化物在苯加氢、费托合成、加氢脱硫/脱氮、醇类分解制氢等精细化工催化反应,和燃料电池电极催化材料、传感器,以及电学材料等领域中有广泛的应用前景。
本发明的机理为:
本发明方法条件温和、环保,突破了传统使用NH3所带来的安全隐患和局限性,通过简单的调控氮源的用量,实现对产物氮化程度的精准控制,及更有效的调节钽的d未占据的空轨道密度,从而使其具有类似贵金属的催化性质。因此本发明方法制备得到的钽基氮氧化物及钽基氮化物在苯加氢、费托合成、加氢脱硫/脱氮、醇类分解制氢等精细化工催化反应,和燃料电池电极催化材料、传感器,以及电学材料等领域中有广泛的应用前景。
本发明相对于现有技术,具有如下的优点及有益效果:
(1)本发明方法产率高,达到95%以上。
(2)本发明制备条件简单易控,工艺条件成本低,制备效率高,产品质量以及成品率高,有良好的应用和产业化前景。
附图说明
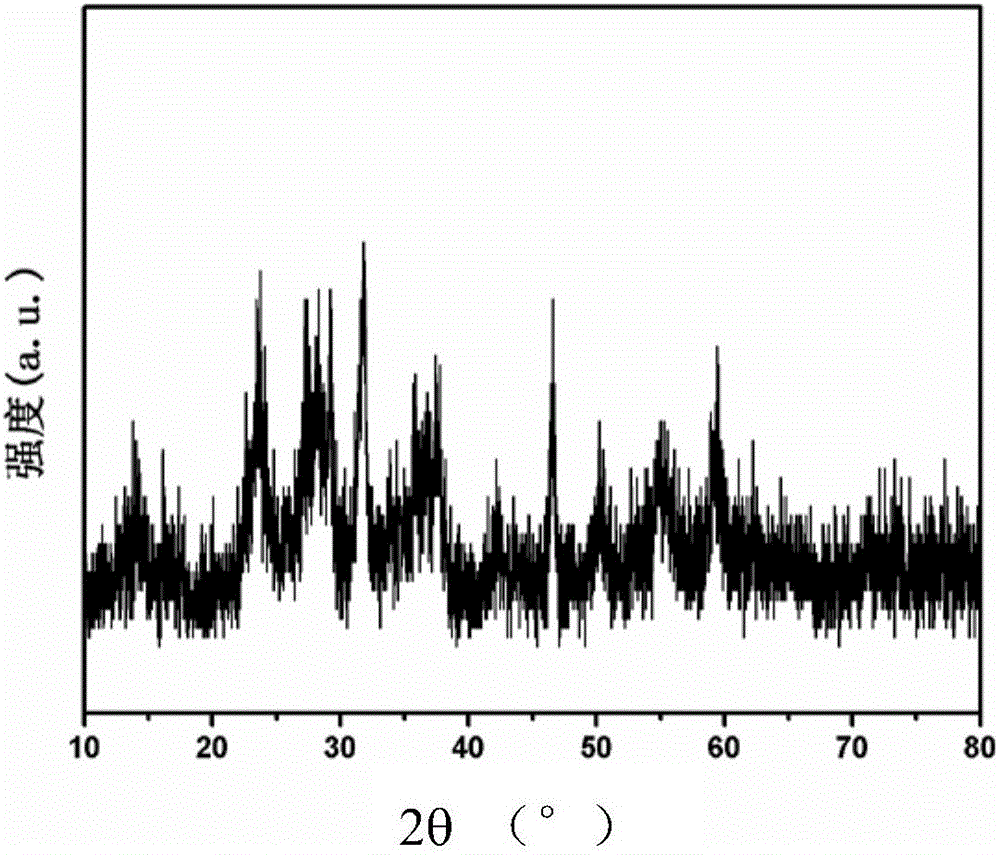
图1是产品A的X射线粉末衍射(XRD)图。
图2是产品A的透射电镜(TEM)图。
图3是产品B的X射线粉末衍射(XRD)图。
图4是产品B的透射电镜(TEM)图。
图5是产品A的扫描电镜(SEM)图。
图6是产品B的扫描电镜(SEM)图。
图7是产品C的扫描电镜(SEM)图。
图8是产品D的扫描电镜(SEM)图。
图9是产品E的扫描电镜(SEM)图。
图10是产品F的扫描电镜(SEM)图。
图11是产品G的扫描电镜(SEM)图。
图12是产品H的扫描电镜(SEM)图。
具体实施方式
下面结合实施例和附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
下列实施例中所用的试剂均可购买得到。
实施例1
将0.25g TaCl5(0.7mM)溶解于2mL甲醇中,随后加入0.103g氯化锶(0.65mM),待溶解后加入0.084g尿素(1.4mM),在室温下搅拌4h至凝胶状。将其在氮气流中700℃焙烧5h,将得到的产物用1mol/L的盐酸处理室温搅拌48h。将得到产物乙醇洗涤数次并抽滤,50℃干燥。得到氮氧化钽纳米颗粒A(TaON)。
实施例2
将0.25g TaCl5(0.7mM)溶解于2mL甲醇中,随后加入0.103g氯化锶(0.65mM),待溶解后加入0.294g尿素(4.9mM),在室温下搅拌4h至凝胶状。将其在氮气流中700℃焙烧5h,将得到的产物用1mol/L的盐酸处理室温搅拌48h。将得到产物乙醇洗涤数次并抽滤,50℃干燥。得到五氮化三钽纳米颗粒B(Ta3N5)。
实施例3
将0.25g TaCl5(0.7mM)溶解于2mL甲醇中,随后加入0.103g氯化锶(0.65mM),待溶解后加入0.084g尿素(1.4mM),在室温下搅拌4h至凝胶状。将其在氩气流中700℃焙烧5h,将得到的产物用1mol/L的盐酸处理室温搅拌48h。将得到产物乙醇洗涤数次并抽滤,50℃干燥。得到氮氧化钽纳米颗粒C(TaON)。
实施例4
将0.25g TaCl5(0.7mM)溶解于2mL甲醇中,随后加入0.555g氯化锶 (3.5mM),待溶解后加入0.084g尿素(1.4mM),在室温下搅拌4h至凝胶状。将其在氮气流中700℃焙烧5h,将得到的产物用1mol/L的盐酸处理室温搅拌48h。将得到产物乙醇洗涤数次并抽滤,50℃干燥。得到氮氧化钽纳米颗粒D(TaON)。
实施例5
将0.25g TaCl5(0.7mM)溶解于2mL甲醇中,随后加入0.01g氯化锶(0.07mM),待溶解后加入0.084g尿素(1.4mM),在室温下搅拌4h至凝胶状。将其在氮气流中700℃焙烧5h,将得到的产物用1mol/L的盐酸处理室温搅拌48h。将得到产物乙醇洗涤数次并抽滤,50℃干燥。得到氮氧化钽纳米颗粒E(TaON)。
实施例6
将0.25g TaCl5(0.7mM)溶解于2mL甲醇中,随后加入0.0665g氯化镁(0.7mM),待溶解后加入0.21g尿素(3.5mM),在室温下搅拌4h至凝胶状。将其在氮气流中700℃焙烧5h,将得到的产物用1mol/L的盐酸处理室温搅拌48h。将得到产物乙醇洗涤数次并抽滤,50℃干燥。得到五氮化三钽纳米颗粒F(Ta3N5)。
实施例7
将0.25g TaCl5(0.7mM)溶解于2mL甲醇中,随后加入0.138g碳酸钡(0.7mM),待溶解后加入0.21g尿素(3.5mM),在室温下搅拌4h至凝胶状。将其在氮气流中700℃焙烧5h,将得到的产物用1mol/L的盐酸处理室温搅拌48h。将得到产物乙醇洗涤数次并抽滤,50℃干燥。得到五氮化三钽纳米颗粒G(Ta3N5)。
实施例8
将0.25g TaCl5(0.7mM)溶解于2mL甲醇中,随后加入0.103g氯化锶 (0.65mM),待溶解后加入0.617g三聚氰胺(4.9mM),在室温下搅拌4h至凝胶状。将其在氮气流中700℃焙烧5h,将得到的产物用1mol/L的盐酸处理室温搅拌48h。将得到产物乙醇洗涤数次并抽滤,50℃干燥。得到五氮化三钽纳米颗粒H(Ta3N5)。
实施例9:纳米颗粒的观察
对实施例1~8制备得到的纳米颗粒采用ZEISS ULTRA55仪器上摄取扫描电镜照片(SEM),在JEOL JEM 2100F仪器上摄取透镜照片(TEM),在Bruker D8型X射线衍射仪上进行XRD扫描,结果见图1~12。
由图1~2可见,实施例1成功制备得到纳米粒径为10nm左右的氮氧化钽。且由图5可见,本发明制备得到的氮氧化钽可在乙醇中分散为颗粒状。
图3~4表明实施例2成功制备得到纳米粒径为10nm左右的五氮化三钽。且由图6可见,本发明制备得到的五氮化三钽在乙醇中具有良好的分散性。
图7表明在不同的惰性气体氛围中均可制备得到分散性良好的纳米颗粒。
图8~9表明,通过改变使用的锶原子和钽原子的摩尔比同样可制备得到氮氧化钽纳米颗粒。
图10~11表明,通过改变碱土金属元素,可同样制备得到五氮化三钽纳米颗粒。
图12表明,当氮源改变为三聚氰胺时,可同样制备得到分散性好的五氮化三钽纳米颗粒。
由上可知,本发明方法制备得到的氮氧化钽和五氮化三钽颗粒小,因此,N原子的插入致使金属晶格扩张,金属间距和晶胞常数变大,金属原子间的相互作用力减弱,产生相应的d带收缩,从而导致Fermi能级附近态密度的重新分布,价电子数增加,结构也随之变化。这种调变使过渡金属氮化物兼具共价化合物、离子晶体和过渡金属三种物质的特性,从而表现在催化性质上完全不同于其相应的金属,而具有与Ⅷ族贵金属相似的性质,在肼分解、NH3合成和分解、加氢精制、烃类异构、选择加氢等方面得到广泛的应用。
同时,其具有的半导体的性质决定了它在光催化分解水领域和光降解有机 污染物及无机染料工业的应用价值。基于本产品具有以上的潜在应用价值,并且制备条件简单易控,工艺条件成本低,制备效率高,产品质量以及成品率高,因此本发明制备得到的纳米颗粒具有良好的应用和产业化前景。对该方法系统研究,不仅可以提供新颖的具有类贵金属性能的能源催化转化碳化物催化材料,而且对材料的合成方法学以及催化剂设计具有广泛的意义。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!