一种四元羧酸N,N,N′,N′‑四(4′‑羧基联苯基)‑1,4‑苯二胺的合成方法与流程
本发明涉及有机化学的技术领域,尤其一种四元羧酸n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺的合成方法。
背景技术:
延长的刚性四羧酸类配体广泛应用于mofs材料的合成中,这类材料通常具有良好的孔道结构、大的比表面积,使这类材料在气体分离与储藏、化学传感、催化及药物缓释等不同领域具有潜在应用。由于芳香四羧酸特别是长链的芳香四羧酸具有以下特性而成为制备高维金属有机框架的较好选择:一、可全部或部分去质子化的羧基可使该配体具有更为灵活多样的配位模式;二、两个邻位的羧基距离较近,易于形成分子内氢键;三、由于自身大的空间位阻,苯环与苯环之间有一定的夹角,部分羧基也会和苯环有一定的夹角,可以从不同方向与金属离子配位;四、分子较高的对称性有利于晶体的生长与结构的设计。比如2009年lin(mal.,jina.,xiez.,etal.angew.chem.int.ed.,2009,48:9905-9908)等使用两个四羧酸配体和铜盐在溶剂热条件下成功合成两个金属有机框架,作者使用冷冻干燥法有效增加了化合物的比表面积,同时也增加了氢气的吸附量。使用冷冻干燥法比真空干燥的多一倍的氢吸附量。stylianou等(stylianouk.c.,heckr.,chongs.y.,etal.j.am.chem.soc.,2010,132:4119-4130)报道使用基于芘的四羧酸配体(1,3,6,8-四(4-苯甲酸)芘(tbapy))和硝酸铟合成一个强荧光的金属有机框架1,1展现了良好的热稳定性。实验表明1在471和529nm处有强的荧光。化合物1的荧光寿命达到0.110ms,量子产率为6.7%。延长的刚性四角羧酸配体通常能得到结构新颖、比表面积大的多微孔mofs材料,并能改变微孔结构,使其微孔内电子环境得到优化从而提高上述应用性能,因此,设计合成结构新颖的延长多羧酸配体并应用于mofs材料的合成已成为当今研究的热点之一。然而,大尺寸的刚性多羧酸化合物合成比较困难。因此本发明中所涉及的结构新颖的n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺是一种具有巨大潜在应用价值的配体。目前尚无文献报道该化合物。
技术实现要素:
本发明的目的是提供一种合成成本低、产率高、产品纯度高的新型n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺的合成方法。
本发明的n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺的合成方法,合成步骤为:
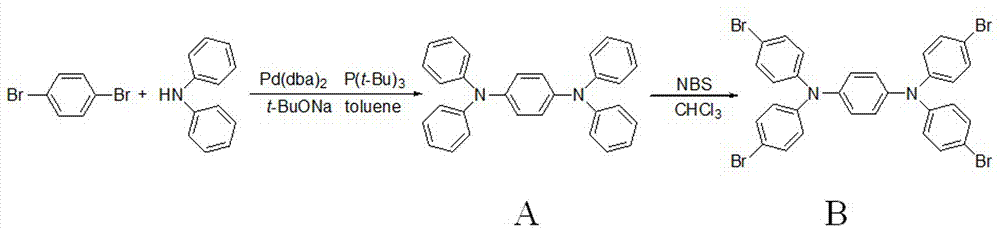
(ⅰ)以1,4-二溴苯和二苯胺为原料,加入一定量叔丁醇钠和适量催化剂,加入甲苯作为溶剂,在氮气或氩气保护下快速加入三-(2-甲苯基)膦,摩尔比1,4-二溴苯:二苯胺:叔丁醇钠为1:2.1~2.3:2.1~2.5,1,4-二溴苯与甲苯的用量比是1毫摩:3.0~4.5毫升。加完后在85~100℃油浴加热反应10~16小时,反应结束蒸出甲苯,加20~40ml水稀释,二氯甲烷萃取(20~40ml×3),收集有机相并用无水亚硫酸钠对其进行除水处理,过滤,真空浓缩,最后以石油醚和乙酸乙酯按体积比为20:0.8~1.2的混合溶剂为洗脱剂用柱色谱法分离提纯得到化合物a:n,n,n′,n′-四苯基-1,4-苯二胺;
(ⅱ)n,n,n′,n′-四苯基-1,4-苯二胺和nbs(n-溴代丁二酰亚胺)按摩尔比1:3.5~5.5混合后溶于一定溶剂中,n,n,n′,n′-四苯基-1,4-苯二胺与溶剂的用量比是1毫摩:6.0~8.0毫升,在0~30℃反应2~10小时,反应结束后加饱和亚硫酸钠溶液10.0~20.0毫升,加水10~30毫升稀释,收集有机相并用无水硫酸镁对其进行除水处理,过滤,真空浓缩,最后用10~30ml无水乙醇重结晶纯化3次,过滤、干燥得化合物b:n,n,n′,n′-四(4-溴苯基)-1,4-苯二胺;
(ⅲ)在氩气或氮气保护下,在250ml三口瓶中分别加入n,n,n′,n′-四(4-溴苯基)-1,4-苯二胺、4-甲氧羰基苯硼酸、碳酸钾和一定量的二氧六环,摩尔比n,n,n′,n′-四(4-溴苯基)-1,4-苯二胺:4-甲氧羰基苯硼酸甲酯:碳酸钾为1:4.5~5.0:10.0~15.0,n,n,n′,n′-四(4-溴苯基)-1,4-苯二胺与溶剂的比为1毫摩:10.0~15.0毫升。加入一定量的钯盐作为催化剂,90℃反应20~30小时,反应结束蒸出1,4-二氧六环,加20~30ml水稀释,二氯甲烷萃取(40~70ml×3),收集有机相并用无水亚硫酸钠对其进行除水处理,过滤,蒸出二氯甲烷溶剂得粗产物,最后以石油醚和二氯甲烷按体积比为4:0.8~1.2的混合溶剂为洗脱剂用柱色谱法分离提纯得到化合物c:n,n,n′,n′-四(4′-甲氧羰基联苯基)-1,4-苯二胺;
(ⅳ)将n,n,n′,n′-四(4′-甲氧羰基联苯基)-1,4-苯二胺和naoh按摩尔比1:30~50加入1,4-二氧六环和水的混合溶剂中,体积比为3~5:1,95℃回流8~12小时,蒸除去过量1,4-二氧六环,加水稀释,加入过量的稀hno3进行酸化至ph=2,析出黄色固体,真空干燥得最终化合物d:n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺。
所述的一种四元羧酸n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺的合成方法,制备a时,在氮气或氩气保护下快速加入三-(2-甲苯基)膦,摩尔比1,4-二溴苯:二苯胺:叔丁醇钠为1:2.1~2.3:2.1~2.5,1,4-二溴苯与甲苯的用量比是1毫摩:3.0~4.5毫升。
所述的一种四元羧酸n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺的合成方法,制备a以甲苯作为溶剂,在85~100℃油浴加热反应10~16小时,反应结束蒸出甲苯,加20~40ml水稀释,二氯甲烷萃取(20~40ml×3),柱层析纯化a以石油醚和乙酸乙酯按体积比为20:0.8~1.2。
所述的一种四元羧酸n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺的合成方法,其特征在于:制备化合物a使用的催化剂为双(二亚苄基丙酮)钯,用量为1,4-二溴苯摩尔数的0.5~1.2%。
所述的一种四元羧酸n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺的合成方法,制备化合物b时,a和nbs的摩尔比为1:3.5~5.5,需加50~80ml二氯甲烷作溶剂。
所述的一种四元羧酸n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺的合成方法,其特征在于:制备化合物b时,纯化b用10~30ml无水乙醇重结晶3次。
所述的一种四元羧酸n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺的合成方法,制备化合物c时,摩尔比b:4-甲氧羰基苯硼酸:碳酸钾为1:4.5~5.0:10.0~15.0。需加80~120ml二氧六环作溶剂。纯化b用石油醚和二氯甲烷按体积比为4:0.8~1.2的混合溶剂为洗脱剂。
所述的一种四元羧酸n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺的合成方法,制备化合物c使用的催化剂为四(三苯基膦)钯,用量为b的物质的量的1~2.5%。
所述的一种四元羧酸n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺的合成方法,n,n,n′,n′-四(4′-甲氧羰基联苯基)-1,4-苯二胺和naoh按摩尔比1:30~50,溶剂1,4-二氧六环和水的混合溶剂中,体积比为3~5:1,酸化至ph=2。
用本方法合成结构新颖的n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺的有益效果是:延长的刚性四羧酸类配体广泛应用于mofs材料的合成中,这类材料通常具有有序的孔道结构、大的比表面积,使这类材料在气体储藏与分离、化学传感、催化及药物缓释等不同领域具有潜在应用。延长的刚性四羧酸配体通常能得到结构新颖、比表面积大的多微孔mofs材料,并能改变微孔结构,使其气孔内电子环境得到优化从而提高上述应用性能,因此,设计合成结构新颖的延长多羧酸配体并应用于mofs材料的合成已成为当今研究的热点之一。然而,大尺寸的刚性多羧酸化合物合成比较困难。因此,用本方法合成结构新颖的n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺是一种具有巨大潜在应用价值的四元羧酸配体。
附图说明
图1本发明中目标化合物d的结构式示意图。
图2本发明中化合物a和b的合成示意图。
图3本发明中化合物c和d的合成示意图。
具体实施方式
本发明的n,n,n′,n′-四(4′-羧基联苯基)-1,4-苯二胺的合成方法,合成步骤为:
以1,4-二溴苯,二苯胺为原料经buchwald-hartwig芳胺化反应生成n,n,n',n'-四苯基-1,4-苯二胺(a),a与nbs溴化反应生成n,n,n',n'-四(4-溴苯基)-1,4-苯二胺(b),合成路线如图2所示。b与对甲氧羰基苯硼酸经suzuki偶联反应生成n,n,n',n'-四(4'-甲氧羰基联苯基)-1,4-苯二胺(c),c最后水解得到目标化合物n,n,n',n'-四(4'-羧基联苯基)-1,4-苯二胺(d),合成路线如图3所示。
具体实施例如下:
(ⅰ)如图2所示:称取1,4-二溴苯(2.9g,12.3mmol),二苯胺(5.0g,29.6mmol),双(二亚苄基酮)钯(0.04g,0.07mmol),叔丁醇钠(2.9g,30.2mmol)于250ml三口瓶中,加入50ml甲苯,通入氩气15min后迅速加入3.5ml三-(2-甲苯基)膦,氩气保护下90℃油浴反应12h,反应结束蒸出甲苯,加30ml水稀释,二氯甲烷萃取(30ml×3),收集有机层,无水na2so3干燥、过滤,蒸出溶剂得反应粗产物,干燥后经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=20:1)纯化,得白色粉末a(3.04g,7.4mmol),产率60%。熔点:197~198℃。
化合物a核磁1hnmr、13cnmr和高分辨质谱数据:1hnmr(400mhz,cdcl3)δ7.28~7.22(m,8h),7.11(m,8h),6.98(m,8h).13cnmr(101mhz,cdcl3)δ142.82,132.12,129.15,125.42,123.70,122.38。hrms(esi),c30h24n2,实测值(计算值),m/z:413.5245[m+h]+(413.5250)。
(ⅱ)如图2所示:称取n,n,n',n'-四苯基-1,4-苯二胺(4.0g,9.7mmol)于250ml三口瓶中,缓慢向三口瓶中加入60ml二氯甲烷,加入nbs(6.9g,38.8mmol),室温搅拌4h,停止反应,加入饱和na2so3溶液,分离有机相,无水mgso4干燥、过滤,蒸出氯仿得反应粗产物,无水乙醇重结晶3次,蒸馏水洗涤沉淀,得白色粉末b(6.9g,9.5mmol),产率,98%。熔点:99~101℃。
化合物b核磁1hnmr、13cnmr和高分辨质谱数据:1hnmr(400mhz,cdcl3)δ7.34(m,4h),7.26(m,4h),7.08(d,j=7.4hz,4h),6.95(m,8h).13cnmr(101mhz,cdcl3)δ:132.31,132.17,131.90,129.40,125.46,125.08。hrms(esi),c30h20br4n2,实测值(计算值),m/z:729.1084[m+h]+(729.1092)。
(ⅲ)如图3所示:在250ml三口瓶中分别加入n,n,n',n'-四(4-溴苯基)-1,4-苯二胺(5.5g,7.6mmol)、4-甲氧羰基苯硼酸(6.6g,4.8eq),k2co3(13.8g,100.32mmol)和100ml1,4-二氧六环,通入n210min后,加2%四(三苯基膦)钯,90℃反应24h,反应结束蒸出1,4-二氧六环,加30ml水稀释,二氯甲烷萃取(60ml×3),收集有机层,无水na2so3干燥、过滤,蒸出二氯甲烷得反应粗产物,干燥后经硅胶柱层析(洗脱剂:石油醚/二氯甲烷=4:1)纯化,得淡黄色粉末n,n,n',n'-四(4'-甲氧羰基联苯基)-1,4-苯二胺(5.2g,5.5mmol),产率72%。熔点:345-347℃。
化合物c核磁1hnmr、13cnmr和高分辨质谱数据:1hnmr(400mhz,cdcl3)δ8.09(d,j=8.4hz,8h),7.99(d,j=8.2hz,8h),7.66(d,j=8.4hz,9h),7.51(d,j=2.1hz,4h),7.35(d,j=8.2hz,9h),3.96(s,12h).13cnmr(101mhz,cdcl3)δ166.97,144.59,142.97,139.63,133.07,131.58,130.15,129.33,129.06,127.69,126.31,117.85,52.27。hrms(esi),c62h48n2o8,实测值(计算值),m/z:959.0536[m+h]+(950.0531)。
(ⅳ)如图3所示:在250ml的圆底烧瓶里依次加入n,n,n',n'-四(4'-甲氧羰基联苯基)-1,4-苯二胺(3.5g,3.7mmol)、naoh(5g,125mmol)、h2o(20ml)和60ml1,4-二氧六环,95℃反应12h,蒸出1,4-二氧六环,加入适量的水溶解羧酸钠盐,过滤,滤液中加稀hno3酸化至ph为2,析出沉淀,静置,倾倒上层清液,抽滤、干燥得目标化合物黄色沉淀d(3.23g,3.6mmol),产率98%。熔点:大于350℃。
化合物d核磁1hnmr、13cnmr和高分辨质谱数据:1hnmr(400mhz,dmso)δ12.96(s,4h),8.11-7.91(m,9h),7.85(m,2h),7.75(m,10h),7.56(m,9h),7.21-6.95(m,6h).13cnmr(101mhz,dmso)δ167.61,131.97,130.42,129.51,129.26,129.14,128.58,127.59,126.62,126.53,124.01。hrms(esi),c58h40n2o8[m-h]+,实测值(计算值),m/z:891.2805(891.2813)。
以上所述,仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,任何未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明技术方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!