还原/pH双重响应性阿霉素前药及其制备方法与应用与流程
本发明属于生物医用高分子材料领域,具体涉及一种基于聚乙二醇的还原/ph双重响应性阿霉素前药、其制备方法及其应用。
背景技术:
癌症是机体在多种因素、多个阶段和多次突变而导致正常细胞癌变的结果,在医学上也称之为恶性肿瘤。癌症的杀伤力大、易转移,通过破坏组织和器官的各项功能,最终导致人们因器官功能的衰竭而死亡。据统计,随着人口的增长,癌症的发病率也在不断增长,严重危害着人类的健康,因此,癌症治疗方法备受国内外医学研究者们的重视。
在过去的几十年里,随着医疗技术的进步,各种癌症的治疗方法得到了迅速发展,包括手术治疗、化学疗法、放射疗法、免疫治疗、激素治疗、靶向治疗等。但是,化疗过程中使用的小分子药物(如:紫杉醇,阿霉素,喜树碱等),在杀死肿瘤细胞作用的同时对正常细胞也有很强的损伤作用,因此,会引起严重的呕吐、头晕、机体机能下降等副作用。同时,小分子药物在血液循环过程中易被机体识别并排出,加上肿瘤组织附近特殊的环境,如错综复杂的血管网络结构、致密的间质结构以及较高的间质液压力等,使得药物很难到达肿瘤细胞内。正是由于这些限制条件影响了抗肿瘤药物在临床上的疗效。
通过前药化,可以有效地解决上述问题。前药(prodrug),也称药物前体、前驱药物,是指经过生物体转化后具有药理作用的化合物。聚合物前药是指具有活性的药物与起运输作用的聚合物载体通过共价键结合,使药物不具有活性。但是,当前药进入机体循环和代谢过程中,可通过水解作用卸除载体,释放出活性药物,发挥药理作用。这种方式可以有效地提高药物的利用率,增强靶向性,降低药物的毒副作用。
在聚合物前药结构中,对于聚合物基底材料的选择至关重要,生物相容性是材料用于药物载体的最基本要求。聚乙二醇(peg)具有无毒性、良好生物相容性和生物可降解性,已被美国食品药品监督管理局(fda)批准使用。通过对peg进行末端官能化修饰,再与疏水性药物键合,可以制备聚合物前药,赋予药物分子一些优良性质。尽管聚合物前药在癌症治疗中具有很大的优势,但是,如何将更多药物传递到肿瘤细胞内,仍然是一直备受关注的问题。药物到达病灶位点需要克服各种屏障,应考虑在血液循环过程中如何保持输运载体的稳定性,延长循环时间;如何高效地进入细胞内,更多地累积在特定的部位;如何对药物可控释放等等。如果不解决这些问题,就会大大地降低药物对癌症的治疗效果。
现有技术中,已有一些关于还原敏感和酸敏感性前药的报道。但是,作为抗肿瘤前药,应当具有良好的生物相容性和生物可降解性,并且,作为抗肿瘤前药,还应当具有下列特征:在水溶液中可以形成稳定的聚合物胶束,亲水性外壳起到稳定胶束、提高胶束血液循环时间的作用;前药胶束在体内循环时,具有抗凝血和抗蛋白吸附的性能;当载药胶束到达肿瘤或病变组织时,能够利用局部组织低的ph条件和高的谷胱甘肽浓度的特点,破坏胶束,快速释放出抗癌药物。因此,需要寻求更多的药效明显、且在肿瘤细胞微环境中具有还原和酸敏感性抗肿瘤前药。
技术实现要素:
本发明的目的是提供一种基于聚乙二醇的还原/ph双重响应性阿霉素前药及其制备方法;该阿霉素前药具有良好的生物相容性和抑制肿瘤细胞增殖的能力。
本发明具体的技术方案为:一种还原/ph双重响应性阿霉素前药,由下列化学结构式表达:
m为3~113,n为3~15。
上述技术方案中,阿霉素前药具有酸敏感性的基团以及还原敏感性的基团;该具有双重响应性的阿霉素前药在结构上含有的疏水性阿霉素部分,用于形成胶束的内核,peg亲水性部分包裹在内核外边形成胶束的外壳,对胶束的稳定性具有很好的作用。在还原和ph条件下,酸敏感基团以及还原敏感基团发生断裂,胶束被破坏,从而快速地释放出聚集在胶束内部的疏水性抗癌药物。
优选的技术方案中,还原/ph双重响应性阿霉素前药的数均分子量为4000~80000g·mol-1。
本发明采用结构中含有酸敏感基团以及还原敏感基团的交替共聚物poly(ss-alt-a)作为基本原料,经铜盐及配体催化化学反应活化后与抗癌药物阿霉素反应,制备基于聚乙二醇的还原/ph双重响应性阿霉素前药dox-hyd-poly(ss-alt-a)-hyd-dox。
上述还原/ph双重响应性阿霉素前药的制备方法,包括以下步骤:
(1)以6-叠氮己酰肼与阿霉素盐酸盐(dox×hcl)作为原料,无水甲醇为溶剂,冰醋酸为催化剂,通过化学反应获得阿霉素衍生物;
其中,6-叠氮己酰肼与阿霉素盐酸盐的摩尔比为1:(2~6);
所述6-叠氮己酰肼的化学结构式为:
所述阿霉素衍生物的化学结构式为:
(2)惰性气体气氛条件下,在铜盐催化剂和配体的存在下,以3,3’-二硫二炔丁基二丙酸酯与二叠氮乙基二缩醛基聚乙二醇为原料,以无水n,n-二甲基甲酰胺为溶剂,通过反应制备获得两端炔基封端的聚乙二醇交替共聚物;
其中,二叠氮乙基二缩醛基聚乙二醇、3,3’-二硫二炔丁基二丙酸酯和铜盐催化剂的摩尔比为1∶(1.1~1.5)∶(0.5~1.5);铜盐催化剂和配体的摩尔比为1∶(1~2);
所述3,3’-二硫二炔丁基二丙酸酯的化学结构式为:
所述二叠氮乙基二缩醛基聚乙二醇的结构式为:
上述m为3~113;
所述两端炔基封端的聚乙二醇交替共聚物的结构式为:
上述m为3~113,n为3~15。
(3)以无水n,n-二甲基甲酰胺作为溶剂,在铜盐催化剂和配体的存在下,用步骤(2)获得的两端炔基封端的的聚乙二醇交替共聚物与步骤(1)获得的阿霉素衍生物反应,制备得到所述还原/ph双重响应性阿霉素前药;
所述两端炔基封端的聚乙二醇交替共聚物、阿霉素衍生物的摩尔比为1:(2~5)。
上述技术方案中,制备阿霉素衍生物(n3-hyd-dox×hcl),主要通过三个步骤制备而成:
第一步,制备6-叠氮己酸甲酯。以6-溴己酸甲酯与叠氮化钠(nan3)反应,以dmf为溶剂,进行叠氮化反应,获得6-叠氮己酸甲酯。其中,6-溴己酸甲酯、叠氮化钠的摩尔比为1:(1.5~5);
第二步,制备6-叠氮己酰肼。以第一步合成的6-叠氮己酸甲酯与质量分数为85%的水合肼反应,以四氢呋喃(thf)作为溶剂,通过酰胺化反应,获得6-叠氮己酰肼。其中,6-叠氮己酸甲酯与水合肼的摩尔比为1:(10~30);
第三步,制备阿霉素衍生物(n3-hyd-dox×hcl)。采用第二步合成的6-叠氮己酰肼与阿霉素盐酸盐(dox×hcl)作为原料,以无水甲醇为溶剂,冰醋酸为催化剂,获得阿霉素衍生物。其中,6-叠氮己酰肼与阿霉素盐酸盐的摩尔比为1:(2~6);
制备两端炔基封端的聚乙二醇交替共聚物包括以下几步:
制备炔基封端的并含有二硫键的小分子化合物3,3’-二硫二炔丁基二丙酸酯(b-ss-b)。采用3,3’-二硫代二丙酸与3-丁炔-1-醇为原料,以1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐为活化剂,4-二甲氨基吡啶为催化剂,二氯甲烷为溶剂,通过酯化反应,获得3,3’-二硫二炔丁基二丙酸酯。其中,3,3’-二硫代二丙酸与3-丁炔-1-醇的摩尔比为1:(1.5~3);
制备叠氮基封端的二叠氮乙基二缩醛基聚乙二醇(n3-a-peg-a-n3),通过两步进行:
第一步,合成二氯乙基二缩醛基聚乙二醇(cl-a-peg-a-cl)。采用聚乙二醇(ho-peg-oh)与2-氯乙基乙烯基醚(ceve)为原料、酸催化剂存在下,反应得到末端为氯基的聚乙二醇产物cl-a-peg-a-cl。其中,聚乙二醇的分子量为:400~5000g·mol-1,聚乙二醇与2-氯乙基乙烯基醚的摩尔比为1:(2~6);酸催化剂为对甲基苯磺酸、对甲苯磺酸吡啶鎓、三氟乙酸中的一种;
第二步,合成二叠氮乙基二缩醛基聚乙二醇(n3-a-peg-a-n3)。将第一步得到的cl-a-peg-a-cl与叠氮化钠反应,以二氯甲烷为溶剂,得到二叠氮乙基二缩醛基聚乙二醇(n3-a-peg-a-n3)。其中,所述末端含氯基的聚乙二醇(cl-a-peg-a-cl)与叠氮化钠的摩尔比为1:(2~6);
制备炔基封端的聚合物poly(ss-alt-a)。在氮气保护下,在铜盐催化剂和配体的存在下,利用上述合成的3,3’-二硫二炔丁基二丙酸酯(b-ss-b)与二叠氮乙基二缩醛基聚乙二醇(n3-a-peg-a-n3)发生化学反应,制备炔基封端的聚合物poly(ss-alt-a)。其中,二叠氮乙基二缩醛基聚乙二醇(n3-a-peg-a-n3)与3,3’-二硫二炔丁基二丙酸酯(b-ss-b)的摩尔比为1:(1.1~1.5);
制备双重响应的阿霉素前药dox-hyd-poly(ss-alt-a)-hyd-dox包括以下几步:
利用脱盐处理的阿霉素衍生物与上步合成的两端炔基封端的聚乙二醇交替共聚物poly(ss-alt-a)发生化学反应,获得还原/ph双重响应性阿霉素前药dox-hyd-poly(ss-alt-a)-hyd-dox。其中,两端炔基封端的聚乙二醇交替共聚物与阿霉素衍生物的摩尔比为1:(2~5)。
上述技术方案中,步骤(1)中,反应温度为60℃~80℃,反应时间为12h~24h;步骤(2)中,惰性气体为氮气;反应温度为25℃~50℃,反应时间为24h~72h;步骤(3)中,反应温度为25℃~50℃,反应时间为24h~48h。
上述技术方案中,步骤(2)和步骤(3)中,铜盐催化剂选自五水合硫酸铜、氯化亚铜或溴化亚铜;配体选自:抗坏血酸钠、联吡啶、五甲基二亚乙基三胺、四甲基乙二胺或六甲基三亚乙基四胺中的一种。
本发明首次在铜盐催化剂和配体的存在下,通过原料以及参数的限定,制备得到含有酸敏感基团以及还原敏感基团的聚乙二醇交替共聚物,解决了现有反应体系无法制备出同时含有酸敏感基团以及还原敏感基团的交替共聚物的问题,是一种新型、简单、高效、快速的合成方法。
进一步的技术方案,在步骤(1)~(3)完成后,分别对产物进行提纯处理,所述纯化过程包括以下步骤:
(i)阿霉素衍生物n3-hyd-dox×hcl的纯化:反应结束后,反应液用棉花过滤除去沉淀物,除去溶剂后在冰乙醚中沉淀三次;采用离心法得到沉淀物,将其放置在真空干燥箱中干燥得到红色粉末状的产物n3-hyd-dox×hcl;
(ii)两端炔基封端的聚乙二醇交替共聚物poly(ss-alt-a)的纯化:反应结束后,用dmf透析48h后,置于60℃油浴中,减压除去dmf;然后用150mlch2cl2重新溶解,用饱和nacl水溶液进行萃取,除去铜盐;有机层用无水na2so4干燥4h后,旋转蒸发除去溶剂,再在乙醚溶液中沉淀3次,将沉淀物收集并真空干燥,最终得到粘性产物poly(ss-alt-a);
(ⅲ)还原/ph双重响应性阿霉素前药dox-hyd-poly(ss-alt-a)-hyd-dox的纯化:反应结束后,采用去离子水透析72h,将透析袋中所得到的红色透明液体冷冻干燥,获得深红色固体产物dox-hyd-poly(ss-alt-a)-hyd-dox。
上述技术方案中,步骤(i)中,采用旋转蒸发的方法除去溶剂;步骤(ii)中透析时采用截留分子量为1000~14000da的透析袋;步骤(ⅲ)中,透析时采用截留分子量为3500~14000da的透析袋。
具体的提纯包括以下步骤:
(i)制备阿霉素衍生物(n3-hyd-dox×hcl)的纯化:
第一步,制备6-叠氮己酸甲酯的纯化:反应结束后,用中性三氧化二铝的短柱过滤反应液,并用油泵减压除去溶剂dmf。随后,用大量的二氯甲烷溶解粗产物,用30ml的二次水萃取三次,将有机相收集并用无水硫酸钠干燥4h。最后,用棉花过滤、旋转蒸发除去溶剂,将产物置于真空烘箱中干燥24h,得到无色的液体产物6-叠氮己酸甲酯;
第二步,制备6-叠氮己酰肼的纯化:反应结束后,旋转蒸发除去thf后换用ch2cl2溶解。在分液漏斗中,采用饱和氯化钠水溶液萃取粗产物。将有机相用无水na2so4干燥4h,除去溶剂后真空干燥,最终收集到产物6-叠氮己酰肼;
第三步,制备阿霉素衍生物的纯化:反应结束后,用棉花过滤除去反应液中的na2so4,将滤液旋转蒸发除去溶剂后,在冰乙醚中沉淀三次。最终,采用离心法得到下面的沉淀物,将其放置在真空干燥箱中干燥24h,得到红色粉末状产物,即为n3-hyd-dox×hcl;
(ii)制备炔基封端的3,3’-二硫二炔丁基二丙酸酯(b-ss-b)的纯化:反应结束后,用滤纸过滤,除去反应中生成的盐,再用150ml的ch2cl2溶解,用1mhcl溶液和饱和氯化钠(nacl)水溶液的混合溶液萃取2次,每个水层继续用30ml的ch2cl2萃取3次。将有机层收集后,用无水na2so4干燥4h,除去溶剂后的粗产物继续采用柱层析法进行分离提纯。收集最终产物,真空干燥24h,即得到3,3’-二硫二炔丁基二丙酸酯(b-ss-b);
(ⅲ)制备叠氮基封端的二叠氮乙基二缩醛基聚乙二醇(n3-a-peg-a-n3)的纯化:
第一步,制备合成二氯乙基二缩醛基聚乙二醇(cl-a-peg-a-cl)的纯化:反应结束后,先用ch2cl2稀释反应液,用10ml的磷酸缓冲溶液(pbs,ph10.0)和饱和nacl水溶液的混合溶液萃取,得到的水层继续用ch2cl2萃取三次。有机层用无水na2so4干燥4h后,旋转蒸发除去大量溶剂,剩约3ml粗产物溶液时,将其在正己烷中沉淀三次,收集沉淀物并真空干燥,得到产物cl-a-peg-a-cl;
第二步,制备合成二叠氮乙基二缩醛基聚乙二醇(n3-a-peg-a-n3)的纯化:反应结束后,用中性al2o3的短柱过滤除去未反应的nan3,60°c油浴减压条件下除去溶剂dmf。随后,用ch2cl2溶解粗产物,用ph10.0的pb缓冲溶液萃取提纯产物,每个水层继续用20ml的ch2cl2萃取,最终将有机相收集并用无水na2so4干燥,旋转蒸发除去溶剂并真空干燥得到棕色的产物n3-a-peg-a-n3;
(ⅳ)两端炔基封端的聚乙二醇交替共聚物poly(ss-alt-a)的纯化:反应结束后,用dmf透析48h后,置于60℃油浴中,减压除去dmf。然后用150mlch2cl2重新溶解,用饱和nacl水溶液进行萃取,除去铜盐。有机层用无水na2so4干燥4h后,旋转蒸发除去溶剂,再在乙醚中沉淀3次,将沉淀物收集并真空干燥,最终得到粘性产物poly(ss-alt-a);
(ⅴ)制备双重响应的阿霉素前药dox-hyd-poly(ss-alt-a)-hyd-dox的纯化:反应结束后,采用去离子水透析72h,将透析袋中所得到的红色透明液体冷冻干燥,得到红色粘性固体产物dox-hyd-poly(ss-alt-a)-hyd-dox。
本发明公开的还原/ph双重响应性阿霉素前药dox-hyd-poly(ss-alt-a)-hyd-dox可以在水溶液中自组装形成前药胶束,疏水性药物阿霉素形成胶束的核;亲水性peg链段形成胶束的外壳,起到稳定胶束的作用。而疏水性内核和亲水性peg链段之间含有还原响应的二硫键和酸敏感的缩醛及酰腙键基团,在还原和ph条件下,敏感基团发生断裂,胶束被破坏,从而快速地释放出聚集在胶束内部的疏水性抗癌药物。因此,本发明同时请求保护上述还原/ph双重响应性阿霉素前药在制备刺激响应性抗癌纳米药物中的应用。
由于上述方案的实施,本发明与现有技术相比,具有以下优点:
1、本发明采用生物相容性良好的聚乙二醇和疏水性抗癌药物阿霉素分别作为亲水性链段和疏水性末端,以在还原条件下和酸性条件下可发生水解的基团作为敏感性连接基团,制备了具有良好生物相容性的还原/ph双重响应性阿霉素前药;与原药阿霉素相比,前药具有良好的水溶性和贮存稳定性,在肿瘤细胞环境中易于分解,加速药物释放,大大增加了其应用价值。
2、本发明获得的还原/ph双重响应性阿霉素前药在水中可自组装形成稳定的聚合物前药胶束。疏水性的阿霉素部分聚集形成胶束内核,亲水性peg链段作为胶束外壳起到稳定胶束、提高胶束血液循环时间的作用。亲水部分和疏水部分通过在酸性条件下可裂解化学键相连,同时亲水的peg链段内部含有在还原条件下和酸性条件下可发生水解的基团;当前药胶束到达肿瘤或病变组织时,由于这些组织微环境的ph值降低,会使酸敏感基团发生断裂,同时在高的谷胱甘肽的环境中,还原敏感基团也发生断裂,导致胶束结构被破坏,从而快速释放出抗癌药物,增加了药物的利用率和靶向性,在癌症的治疗方面,具有潜在的应用价值。
3、本发明提供的还原/ph双重响应性阿霉素前药结构明确,合成条件不苛刻,具有以下显著特点:(1)所用原料和试剂容易获得;(2)反应条件简单;(3)产率高;(4)立体选择性好;(5)产物分离简便;(6)产物稳定性好;制备容易,提纯方便,适合工业化生产。
附图说明
图1为实施例一中阿霉素衍生物(n3-hyd-dox×hcl)的核磁共振氢谱图,溶剂为氘代二甲亚砜;
图2为实施例二中3,3’-二硫二炔丁基二丙酸酯的核磁共振氢谱图,溶剂为氘代氯仿;
图3为实施例三中cl-a-peg21-a-cl和n3-a-peg21-a-n3的核磁共振氢谱图,溶剂为氘代氯仿,其中,下标21代表-ch2ch2o-的重复单元数;
图4为实施例四中聚合物poly(ss-alt-a)3.6的核磁共振氢谱图,溶剂为氘代氯仿,其中,下标3.6代表交替共聚物的聚合度;
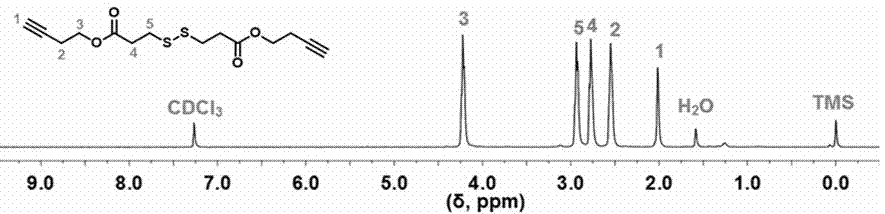
图5为实施例五中dox-hyd-poly(ss-alt-a)3.6-hyd-dox的核磁共振氢谱图,溶剂为氘代氯仿;
图6为实施例一~五中ho-peg21-oh,cl-a-peg21-a-cl,n3-a-peg21-a-n3,poly(ss-alt-a)3.6,n3-hyd-dox×hcl和dox-hyd-poly(ss-alt-a)3.6-hyd-dox的红外光谱图;
图7为实施例五中dox-hyd-poly(ss-alt-a)3.6-hyd-dox的在ph7.4缓冲溶液中自组装形成的胶束的动态光散射曲线和透射电镜照片图;
图8为实施例五中dox-hyd-poly(ss-alt-a)3.6-hyd-dox形成的聚合物前药胶束与裸药阿霉素在不同ph值的缓冲溶液中的药物释放曲线图;
图9为dox-hyd-poly(ss-alt-a)3.6-hyd-dox前药与裸药阿霉素的抑制肿瘤细胞增殖性能测试图。
具体实施方式
下面结合实施例和附图对本发明作进一步描述:
实施例一:阿霉素衍生物(n3-hyd-dox×hcl)的合成
阿霉素衍生物(n3-hyd-dox×hcl)主要通过三个步骤制备而成。第一步,制备6-叠氮己酸甲酯;第二步,制备6-叠氮己酰肼;第三步,对阿霉素盐酸盐(dox×hcl)进行修饰得到阿霉素衍生物(n3-hyd-dox×hcl),具体合成方法如下:
合成小分子化合物6-叠氮己酸甲酯。将事先放置于120°c烘箱的装置取出,放入保干器中,待其冷却至室温,取出使用。分别称取6-溴己酸甲酯(3.54g,16.93mmol)和叠氮化钠(nan3)(2.75g,42.33mmol),并用20ml的dmf溶解在50ml的圆底烧瓶中,在60°c冷凝水回流条件下反应12h后结束。用中性三氧化二铝(al2o3)的短柱过滤反应液,除去未反应的nan3,并用油泵减压除去溶剂dmf。随后,用大量的二氯甲烷(ch2cl2)溶解粗产物,用30ml的二次水萃取三次,将有机相收集并用无水硫酸钠(na2so4)干燥4h。最后,用棉花过滤、旋转蒸发除去溶剂,将产物置于真空烘箱中干燥24h,得到无色的液体产物6-叠氮己酸甲酯(2.47g,产率:53.2%);
在圆底烧瓶中加入已得到的6-叠氮己酸甲酯(0.76g,4.44mmol)和质量分数为85%的水合肼(5.60g,111.1mmol),采用四氢呋喃(thf)作为溶剂。同样,在80°c油浴并装有蛇形冷凝管的装置中,回流反应12h。反应结束后,旋转蒸发除去thf,用ch2cl2溶解。在分液漏斗中,采用饱和氯化钠水溶液萃取粗产物。将有机相用无水na2so4干燥4h,除去溶剂后真空干燥,最终收集到产物6-叠氮己酰肼(0.55g,产率:56.2%);
分别称取6-叠氮己酰肼(102.0mg,0.60mmol),dox×hcl(115.9mg,0.20mmol)和无水na2so4(103mg,0.73mmol)加入带有冷凝回流装置的圆底烧瓶中,用30ml的无水甲醇溶解。随后加入3滴冰醋酸,充分搅拌均匀后,在65°c油浴中避光反应24h。反应结束后,用棉花过滤,除去反应液中的na2so4,除去溶剂后在冰乙醚中沉淀三次。最终,采用离心法得到下面的沉淀物,将其放置在真空干燥箱中干燥24h,得到红色粉末状的产物,即为n3-hyd-dox×hcl(93.5mg,产率:62.4%)。
实施例二:炔基封端的3,3’-二硫二炔丁基二丙酸酯(b-ss-b)的合成
首先,以3,3’-二硫代二丙酸与3-丁炔-1-醇为原料,以1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐为活化剂,4-二甲氨基吡啶为催化剂,二氯甲烷为溶剂,通过酯化反应,获得3,3’-二硫二炔丁基二丙酸酯。具体合成方法如下:
将所用玻璃仪器置于120°c烘箱中烘干4h,取出放在保干器中冷却至室温待用。首先,向三颈烧瓶中依次加入3,3’-二硫代二丙酸(3.66g,17.40mmol)、1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edc×hcl)(8.67g,45.23mmol)和4-二甲氨基吡啶(dmap)(3.19g,2.61mmol),用100ml的ch2cl2溶解;其次,将3-丁炔-1-醇(3.17g,45.23mmol)和15ml的ch2cl2加入恒压滴液漏斗中。在冰水浴中滴加2h,滴加结束后,将装置移到25°c的油浴中,继续反应48h;
反应结束后,旋转蒸发除去少量溶剂,滤纸过滤除去反应生成的盐,再用150ml的ch2cl2溶解,用1mhcl溶液和饱和氯化钠(nacl)水溶液的混合溶液萃取2次,收集的水层用30ml的ch2cl2萃取3次。将有机层收集后,用无水na2so4干燥4h,除去溶剂后的粗产物继续采用柱层析法进行分离提纯,淋洗剂为乙酸乙酯和石油醚的混合溶液(乙酸乙酯:石油醚=1:6)。最终产物被收集,真空干燥24h后即得到较纯的3,3’-二硫二炔丁基二丙酸酯b-ss-b(3.63g,产率:59.4%)。
实施例三:叠氮基封端的二叠氮乙基二缩醛基聚乙二醇(n3-a-peg21-a-n3)的合成
叠氮基封端的化合物二叠氮乙基二缩醛基聚乙二醇(n3-a-peg21-a-n3),通过两步反应得到:首先,合成二氯乙基二缩醛基聚乙二醇(cl-a-peg21-a-cl);其次,合成二叠氮乙基二缩醛基聚乙二醇(n3-a-peg21-a-n3)。具体合成方法如下:
第一步,合成二氯乙基二缩醛基聚乙二醇(cl-a-peg21-a-cl)。取分子量为1000的聚乙二醇(ho-peg22-oh)(6.01g,6.01mmol)和对甲苯磺酸吡啶鎓(ppts)(0.30g,1.20mmol)置于100ml的支管烧瓶中,并加入50ml的甲苯,采用两次常压甲苯共沸的方法除去杂质水。待其冷却后,在氮气保护下,加入30ml干燥的ch2cl2溶解,并在恒压滴液漏斗中加入10ml的ch2cl2和2-氯乙基乙烯基醚(ceve)(3.20g,30.05mmol),冰水浴滴加完后,室温下继续反应1h,加入10ml的质量分数为5%的碳酸钠(na2co3)水溶液终止反应;
产物的提纯采用萃取和沉淀相结合的方法。先用ch2cl2稀释反应液,用10ml磷酸缓冲溶液(pbs,ph10.0)和饱和nacl水溶液的混合溶液萃取,得到的水层继续用ch2cl2萃取三次。有机层用无水na2so4干燥4h后,旋转蒸发除去ch2cl2溶剂。然后,将其在正己烷中沉淀三次,收集沉淀物并真空干燥,得到产物cl-a-peg21-a-cl(5.75g,产率:78.9%);
第二步,合成二叠氮乙基二缩醛基聚乙二醇(n3-a-peg21-a-n3)。将上步得到的cl-a-peg-a-cl(2.37g,1.95mmol)和nan3(0.76g,11.70mmol)溶解在25ml的dmf中,60°c油浴下反应48h。反应结束后,用中性al2o3的短柱过滤除去未反应的nan3,置于60℃油浴中,减压除去dmf。随后,用ch2cl2溶解粗产物,用ph10.0的pb缓冲溶液萃取提纯产物,收集的水层用20ml的ch2cl2萃取。最终将有机相收集,并用无水na2so4干燥,旋转蒸发除去溶剂,真空干燥得到棕色的产物n3-a-peg21-a-n3(1.68g,产率:63.9%)。
实施例四:炔基封端的聚合物poly(ss-alt-a)n的合成
在氮气保护下,将3,3’-二硫二炔丁基二丙酸酯(b-ss-b)与二叠氮乙基二缩醛基聚乙二醇(n3-a-peg21-a-n3)发生化学反应,制备炔基封端的聚合物poly(ss-alt-a)n。具体合成方法如下:
在氮气的保护下,向含有10ml的dmf的支管烧瓶中加入cubr(0.085g,0.59mmol)和五甲基二乙烯三胺(pmdeta)(0.102g,0.59mmol),搅拌使两者进行络合,随后依次加入已合成的b-ss-b(0.25g,0.79mmol)和n3-a-peg21-a-n3(0.97g,0.79mmol)。抽充气三次后,装置在氮气保护下密封好置于50°c油浴中反应48h,随后,补加10mol%的b-ss-b继续反应24h,以确保两端为丁炔基。粗产物用dmf进行透析(透析袋截留分子量为3500da),每隔一段时间换新的透析液。48h后,60°c油泵减压除去dmf。然后用150ml的ch2cl2重新溶解,加入饱和nacl水溶液除去铜盐。分离得到的有机层用无水na2so4干燥4h后,旋转蒸发除去溶剂,再在乙醚溶液中沉淀3次,将沉淀物收集并真空干燥,最终得到粘性产物poly(ss-alt-a)n(0.74g,产率:60.7%)。
实施例五:双重响应的阿霉素前药dox-hyd-poly(ss-alt-a)n-hyd-dox的合成
利用脱盐处理的阿霉素衍生物与聚合物poly(ss-alt-a)n发生化学反应,获得还原/ph双重响应性阿霉素前药dox-hyd-poly(ss-alt-a)n-hyd-dox。具体合成方法如下:
首先,对阿霉素衍生物(n3-hyd-dox×hcl)进行脱盐酸处理。在圆底烧瓶中,加入n3-hyd-dox×hcl(0.095g,0.13mmol),用3mldmso溶解,并加入10ml三乙胺(tea)搅拌2h,静置后吸走上清液再加入新的tea溶液继续搅拌,如此重复2次,最后留取dmso层的样品待用。在支管烧瓶中,通氮气的状态下,先加入cubr(0.015g,0.104mmol)和pmdeta(0.018g,0.104mmol),用5mldmf溶剂溶解并搅拌5min,再依次向瓶中加入poly(ss-alt-a)n(0.23g,0.052mmol)和上述脱去盐酸盐的n3-hyd-dox的溶液,抽充气三次后,将瓶子密封置于50°c油浴中反应48h。反应结束后,将反应液移到截留分子量为5000da的透析袋中,采用去离子水进行透析,每隔4h后换一次去离子水。透析72h后,将透析袋中溶液取出,在冰箱中冷冻后置于冻干机中冻干,最后得到红色粘性固体产物dox-hyd-poly(ss-alt-a)n-hyd-dox(0.26g,产率:84.2%)。
分别利用核磁共振氢谱(1hnmr)和红外光谱(ft-ir)验证所得小分子化合物、聚合物以及基于聚乙二醇的还原/ph双重响应性阿霉素前药的结构。附图1为阿霉素衍生物(n3-hyd-dox×hcl)的核磁共振氢谱图、附图2为聚合物3,3’-二硫二炔丁基二丙酸酯的核磁共振氢谱图、附图3为cl-a-peg21-a-cl和n3-a-peg21-a-n3的核磁共振氢谱图、附图4为聚合物poly(ss-alt-a)3.6的核磁共振氢谱图、附图5为dox-hyd-poly(ss-alt-a)3.6-hyd-dox的核磁共振氢谱图、附图6为ho-peg21-oh,cl-a-peg21-a-cl,n3-a-peg21-a-n3,poly(ss-alt-a)3.6,dox-hyd-n3和dox-hyd-poly(ss-alt-a)3.6-hyd-dox的红外光谱图。结合附图1、2、3、4、5、6可以证明反应产物符合实验设计。
实施例六:采用透析法制备聚合物前药胶束
取聚合物前药(dox-hyd-poly(ss-alt-a)3.6-hyd-dox)25mg溶解在2ml的dmf中,搅拌2h后,用微量注射器以2mlh-1的速度向溶液中滴加20ml的去离子水,滴加结束后,继续搅拌2h。随后,将溶液装入截留分子量为5000da的透析袋中,用去离子水透析24h后,将溶液取出并定容至25ml,最终得到浓度为1mgml-1的胶束溶液。附图7为dox-hyd-poly(ss-alt-a)3.6-hyd-dox在水中自组装形成的胶束的透射电镜照片(a)和动态光散射曲线(b);如:附图7(a)所示为聚合物前药在水溶液中自组装形成球形胶束的形貌;附图7(b)所示为对应胶束粒径的动态光散射测试曲线图,可以看出聚合物前药胶束平均粒径为48nm。
实施例七:聚合物前药的体外药物释放测试
取4ml预先制备好的胶束溶液(胶束浓度为1mgml-1)置于截留分子量为7000da的透析袋中,两端封好置于50ml的大离心管中,随后,向相应的离心管中分别加入30ml不同的缓冲溶液。缓冲溶液分为四种:(1)磷酸缓冲液(ph7.4,10mm);(2)含有10mm的gsh的磷酸缓冲液(ph7.4,10mm);(3)醋酸缓冲液(ph5.0,10mm);(4)含有10mm的gsh的醋酸缓冲液(ph5.0,10mm)。然后,将大离心管置于37.5°c的恒温振荡仪中,以160r/min速度进行振荡。在设定的时间点,依次取出5ml的释放液并补加相应的缓冲溶液。每组实验进行3个平衡实验,最后取平均值。取出的释放液用荧光分光光度计对dox浓度进行测定。载有阿霉素的聚合物胶束在不同的还原和ph条件下的释放曲线如附图8所示,在ph5.0或者在含有10mm的gsh的磷酸缓冲液中,药物释放速率明显快于正常生理条件下;同时,在含有10mm的gsh的ph5.0缓冲溶液中药物累计释放率最大。药物释放说明该聚合物前药胶束具有明显的还原和ph双重响应性,可以达到控制释放的目的。
实施例八:聚合物前药胶束抑制肿瘤细胞增殖性能测试
分别将人子宫颈癌细胞(helacells)和人肝癌细胞(hepg2cells)培养在补充有10%胎牛血清(fbs)的dmem培养基中,置于37℃,5%co2(相对湿度为90%)的培养箱中培养,定期更换培养液。选择处在生长活跃期的细胞接种于每孔含有100μldmem培养基的96孔板中,培养24h。用透析法配置聚合物前药dox-hyd-poly(ss-alt-a)3.6-hyd-dox的胶束母液(阿霉素浓度为94mgl-1),将一系列不同浓度的胶束溶液加入到96孔板中,分别继续培养48h和72h。接着加入25μl的mtt试剂,进一步培养4h后,用酶标仪(bio-radmodel680)在570nm下测量对应的吸光度。细胞存活率的计算方法为:细胞存活率(cellviability)(%)=[a]test/[a]control×100%,其中,[a]test为dox-hyd-poly(ss-alt-a)3.6-hyd-dox聚合物前药胶束存在下测得的吸光度,而[a]control为不含聚合物前药的情况下测得的吸光度。每个样品测试三次,取其平均值。如附图9所示,(a)和(b)分别为与hela细胞培养48h和72h后细胞的存活率;(c)和(d)分别为与hepg2细胞培养48h和72h后细胞的存活率。结果显示:与抗癌药物阿霉素相比,所合成的聚合物前药具有良好的抑制hela细胞和hepg2细胞增殖的能力。
本发明首先制备阿霉素衍生物(n3-hyd-dox×hcl);再分别制备炔基封端的小分子化合物3,3’-二硫二炔丁基二丙酸酯(b-ss-b)和叠氮基封端的化合物二叠氮乙基二缩醛基聚乙二醇(n3-a-peg-a-n3);然后,通过聚合反应制备含有缩醛键和二硫键的聚合物poly(ss-alt-a);最后,再通过反应将阿霉素连接在聚合物两端,获得具有还原和ph双重响应的阿霉素前药dox-hyd-poly(ss-alt-a)3.6-hyd-dox,更换本发明限定的其他催化剂以及配体同样可以得到产物,更换聚乙二醇的分子量以及本发明限定的原料比例,可以得到不同m值n值的产物;本发明限定的具有刺激响应性聚合物前药在水溶液中可以组装成前药胶束并稳定存在,而在酸性或者是还原条件下,敏感性基团或链段会发生断裂使胶束被破坏,从而可以将聚集在胶束内部的药物快速地释放出来,用于治疗,取得了如实施例记载的技术效果。
- 还没有人留言评论。精彩留言会获得点赞!