一种1‑吩噻嗪基‑3‑芳基‑3‑(2,4,6‑嘧啶三酮基)‑1‑丙酮及其制备方法与流程
本发明属于化学合成领域,特别涉及一种1-吩噻嗪基-3-芳基-3-(2,4,6-嘧啶三酮基)-1-丙酮及其制备方法。
背景技术:
n-吩噻嗪基查尔酮由于其含有一个a,β-不饱和双键,所以关于n-吩噻嗪基查尔酮的反应多围绕michael加成反应或者合环反应进行。它可与肼类、腈、活泼亚甲基化合物或胺等物质进行michael加成反应,生成一系列含有吩噻嗪基的衍生物,该类衍生物在抗菌,抗病毒等领域有很好的应用。
michael加成反应是一类重要的形成碳—碳键的有机反应,其最直接的应用就是增长碳链,合成含有多个官能团的化合物,这些化合物均具有进一步反应的能力,生成各种类型的有机化合物。但传统的michael反应大多是以强碱作催化剂在有机溶剂中进行,催化剂碱性太强或者反应时间较长,会有很多的副产物产生,从而产率不理想。
技术实现要素:
本发明的目的在于提供一种1-吩噻嗪基-3-芳基-3-(2,4,6-嘧啶三酮基)-1-丙酮及其制备方法,采用固相研磨法,合成了一系列未见文献报道的新的michael加成产物,该方法反应时间短、无须溶剂、绿色、环保、经济、且操作简单、反应条件温和,后处理简单、产率高。
为达到上述目的,本发明采用以下技术方案:
一种1-吩噻嗪基-3-芳基-3-(2,4,6-嘧啶三酮基)-1-丙酮,其结构通式如下:
其中ar为苯基、取代苯基或杂环基。
所述取代苯基为对氯苯基、对溴苯基、对氟苯基、甲基苯基、甲氧基苯基、对硝基苯基、间硝基苯基、邻硝基苯基、二甲氨基苯基、羟基苯基、苯乙烯基;
所述杂环基为呋喃基、噻吩基或吡啶基。
所述的1-吩噻嗪基-3-芳基-3-(2,4,6-嘧啶三酮基)-1-丙酮的制备方法,其具体步骤如下:
向干燥的研钵中加入amol1-吩噻嗪基-3-芳基-丙烯酮,bmol催化剂,cmol巴比妥酸,研磨使其进行反应,tlc跟踪检测直至反应完全,然后水洗、抽滤,将滤饼干燥后即得到1-吩噻嗪基-3-芳基-3-(2,4,6-嘧啶三酮基)-1-丙酮,其中a:b:c=1:(1~1.5):(1~1.5)。
所述1-吩噻嗪基-3-芳基-丙烯酮的结构通式如下:
其中ar为苯基、对氯苯基、对溴苯基、对氟苯基、甲基苯基、甲氧基苯基、对硝基苯基、间硝基苯基、邻硝基苯基、二甲氨基苯基、羟基苯基、苯乙烯基、呋喃基、噻吩基或吡啶基。
所述催化剂为固体naoh或无水k2co3。
所述研磨反应的时间为30~40min。
当1-吩噻嗪基-3-芳基-丙烯酮的原料点消失时表示原料反应完全;tlc检测所用的展开剂为体积比为1:5的乙酸乙酯和石油醚的混合溶剂。
抽滤时,对滤饼进行多次水洗,直到滤液的ph呈中性。
与现有技术相比,本发明具有以下有益效果:
本发明采用固相研磨法,由巴比妥酸和1-吩噻嗪基-3-芳基-丙烯酮反应,合成了一系列未见文献报道的新的michael加成产物,即1-吩噻嗪基-3-芳基-3-(2,4,6-嘧啶三酮基)-1-丙酮,并探索了其制备方法,同时优化了实验条件,缩短了反应时间,为绿色合成做出了贡献。本发明提供的1-吩噻嗪基-3-芳基-3-(2,4,6-嘧啶三酮基)-1-丙酮是一系列全新的含有吩噻嗪基的酮类衍生物,具有杀菌、抑菌等功效,在抗菌、抗病毒、医药等领域有很好的应用前景。
本发明提供的1-吩噻嗪基-3-芳基-3-(2,4,6-嘧啶三酮基)-1-丙酮的制备方法,将1-吩噻嗪基-3-芳基-丙烯酮、催化剂和巴比妥酸加入干燥的研钵中迅速研磨进行反应,研磨过程中,原料随着反应的进行开始变成粘稠状而后逐渐变为固体,tlc跟踪检测至反应完全,产物经水洗、抽滤、干燥即得到1-吩噻嗪基-3-芳基-3-(2,4,6-嘧啶三酮基)-1-丙酮。与传统合成方法相比,本发明采用固相研磨法,反应过程简单,反应现象明显,反应时间短、无须溶剂、绿色、环保、经济、且操作简单、反应条件温和,设备要求低,且该方法的后处理简单、副产物少,产率高。克服了传统合成方法反应时间长,副产物多,产率低等缺点,具有经济、方便、高效、绿色的优点。
进一步的,本发明在制备过程中,用tlc监测反应过程,所用的展开剂为体积比为1:5的乙酸乙酯和石油醚,监测准确,利于控制反应进度和结束。同时本发明所用的催化剂为固体naoh或无水k2co3,廉价易得,催化效果良好,使反应更加迅速和完全,且后处理简单。
附图说明
图1为实施例1制得的产物的红外图谱;
图2为实施例2制得的产物的红外图谱;
图3为实施例3制得的产物的红外图谱;
图4为实施例4制得的产物的红外图谱;
图5为实施例9制得的产物的红外图谱;
图6为实施例10制得的产物的红外图谱;
图7为实施例4制得的产物的1hnmr图谱;
图8为实施例7制得的产物的1hnmr图谱。
具体实施方式
本发明提供的1-吩噻嗪基-3-芳基-3-(2,4,6-嘧啶三酮基)-1-丙酮的反应方程式如下:
其中ar为苯基、对氯苯基、对溴苯基、对氟苯基、甲基苯基、甲氧基苯基、对硝基苯基、间硝基苯基、邻硝基苯基、二甲氨基苯基、羟基苯基、苯乙烯基、呋喃基、噻吩基或吡啶基。
下面将结合本发明较佳的实施例对本发明做进一步详细说明。
实施例1
称取0.0015mol巴比妥酸和0.0013mol无水k2co3于干燥的研钵中,迅速将其混合均匀,再加入0.001mol1-吩噻嗪基-3-苯基-丙烯酮,研磨35min。研磨过程中,混合物会逐渐变黏,继续研磨直至物质不再发生变化,使用tlc跟踪监测反应进程,所用的展开剂为体积比为1:5的乙酸乙酯和石油醚的混合溶剂,待反应结束后,用纯净水对产物进行多次水洗抽滤,直到滤液的ph呈中性,然后将滤饼自然风干,得灰色粉末,即为1-吩噻嗪基-3-苯基-3-(2,4,6-嘧啶三酮基)-1-丙酮。产率为:86.2%,m.p.104℃~107℃。
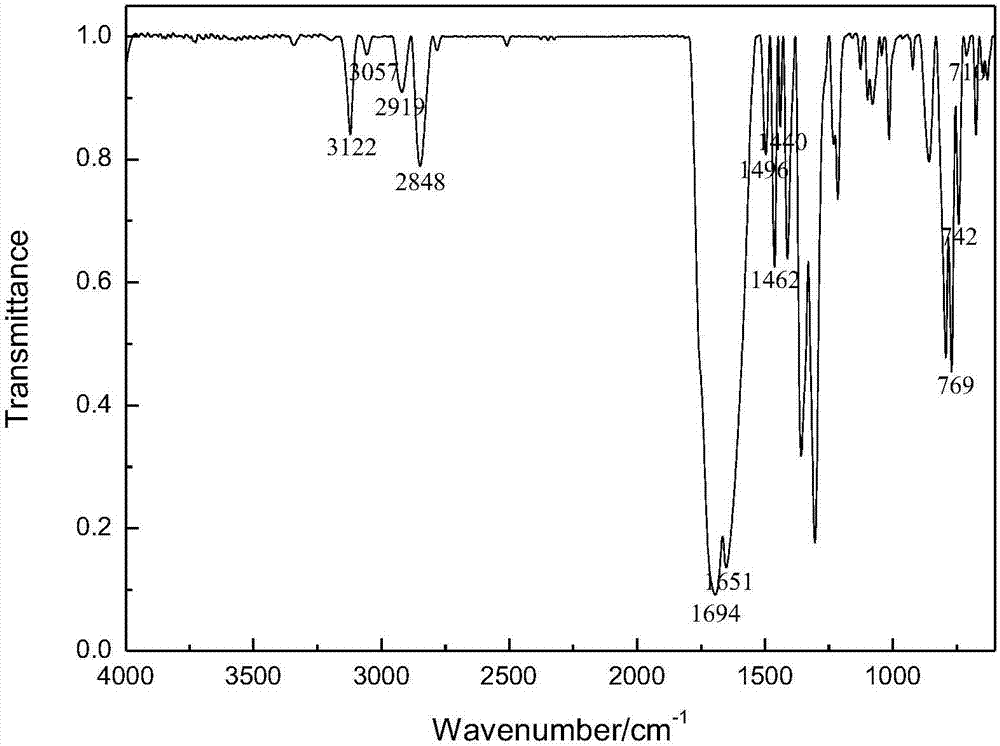
图1是本发明实施例1制备的1-吩噻嗪基-3-苯基-3-(2,4,6-嘧啶三酮基)-1-丙酮的红外图谱,由图1可知:3057cm-1处为ar-h的振动吸收峰,在3122cm-1处为-nh-的吸收峰,在2919cm-1,2848cm-1,1440cm-1处为-ch2的吸收峰,在1694cm-1,1651cm-1处为c=o的特征吸收峰,1496cm-1和1462cm-1处的峰为苯环的呼吸振动吸收峰,742cm-1为苯环上的邻二取代的吸收峰,710cm-1,769cm-1为苯环上单取代的吸收峰。综上所述,可以证实合成得到了1-吩噻嗪基-3-苯基-3-(2,4,6-嘧啶三酮基)-1-丙酮。
1hnmr:6.78-7.22(m,13h,ar-h),10.04(s,1h,-conh),3.94-3.95(d,1h,-cochco-),3.64-3.70(m,1h,-ch),2.63-2.65(d,2h,-coch2).
实施例2
称取0.0015mol巴比妥酸和0.0015mol无水k2co3于干燥的研钵中,迅速将其混合均匀,再加入0.001mol1-吩噻嗪基-3-(4-氯苯基)-丙烯酮,研磨37min。研磨过程中,混合物会逐渐变黏,继续研磨直至物质不再发生变化,使用tlc跟踪监测反应进程,所用的展开剂为体积比为1:5的乙酸乙酯和石油醚的混合溶剂,待反应结束后,用纯净水对产物进行多次水洗抽滤,直到滤液的ph呈中性,然后将滤饼自然风干,得淡黄色粉末,即为1-吩噻嗪基-3-(4-氯苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。产率为:87.4%,m.p.120℃~123℃。
图2是本发明实施例2制备的1-吩噻嗪基-3-(4-氯苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮的红外图谱,由图2可知:3058cm-1处为ar-h的振动吸收峰,在3325cm-1处为-nh-的吸收峰,在2923cm-1,2846cm-1,1439cm-1处为-ch2的吸收峰,在1707cm-1,1674cm-1处为c=o的特征吸收峰,1594cm-1和1464cm-1处的峰为苯环的呼吸振动吸收峰,742cm-1为苯环上的邻二取代的吸收峰,816cm-1为苯环上对位取代的吸收峰。综上所述,可以证实合成得到了1-吩噻嗪基-3-(4-氯苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。
1hnmr:6.65-7.16(m,12h,ar-h),10.24(s,1h,-conh),3.92-3.94(d,1h,-cochco-),3.74-3.78(m,1h,-ch),2.67-2.68(d,2h,-coch2).
实施例3
称取0.0012mol巴比妥酸和0.0015mol无水k2co3于干燥的研钵中,迅速将其混合均匀,再加入0.001mol1-吩噻嗪基-3-(4-溴苯基)-丙烯酮,研磨33min。研磨过程中,混合物会逐渐变黏,继续研磨直至物质不再发生变化,使用tlc跟踪监测反应进程,所用的展开剂为体积比为1:5的乙酸乙酯和石油醚的混合溶剂,待反应结束后,用纯净水对产物进行多次水洗抽滤,直到滤液的ph呈中性,然后将滤饼自然风干,得灰色粉末,即为1-吩噻嗪基-3-(4-溴苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。产率为86.9%,m.p.115℃~118℃。
图3是本发明实施例3制备的1-吩噻嗪基-3-(4-溴苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮的红外图谱,由图3可知:3055cm-1处为ar-h的振动吸收峰,在3328cm-1处为-nh-的吸收峰,在2922cm-1,2847cm-1,1442cm-1处为-ch2的吸收峰,在1710cm-1,1665cm-1处为c=o的特征吸收峰,1598cm-1和1470cm-1处的峰为苯环的呼吸振动吸收峰,738cm-1为苯环上的邻二取代的吸收峰,817cm-1为苯环上对位取代的吸收峰。综上所述,可以证实合成得到了1-吩噻嗪基-3-(4-溴苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。
1hnmr:6.64-7.45(m,12h,ar-h),10.24(s,1h,-conh),3.98-4.02(d,1h,-cochco-),3.72-3.76(m,1h,-ch),2.60-2.62(d,2h,-coch2).
实施例4
称取0.0014mol巴比妥酸和0.0014molnaoh于干燥的研钵中,迅速将其混合均匀,再加入0.001mol1-吩噻嗪基-3-(4-氟苯基)-丙烯酮,研磨34min。研磨过程中,混合物会逐渐变黏,继续研磨直至物质不再发生变化,使用tlc跟踪监测反应进程,所用的展开剂为体积比为1:5的乙酸乙酯和石油醚的混合溶剂,待反应结束后,用纯净水对产物进行多次水洗抽滤,直到滤液的ph呈中性,然后将滤饼自然风干,得灰色粉末,即为1-吩噻嗪基-3-(4-氟苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。产率为87.7%,m.p.115℃~117℃。
图4是本发明实施例4制备的1-吩噻嗪基-3-(4-氟苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮的红外图谱,由图4可知:3057cm-1处为ar-h的振动吸收峰,在3298cm-1处为-nh-的吸收峰,在2927cm-1,2851cm-1,1439cm-1处为-ch2的吸收峰,在1711cm-1,1667cm-1处为c=o的特征吸收峰,1595cm-1和1463cm-1处的峰为苯环的呼吸振动吸收峰,742cm-1为苯环上的邻二取代的吸收峰,817cm-1为苯环上对位取代的吸收峰。综上所述,可以证实合成得到了1-吩噻嗪基-3-(4-氟苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。
图7是本发明实施例4制备的1-吩噻嗪基-3-(4-氟苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮的1hnmr图谱,由图7可知:1hnmr:6.64-7.12(m,12h,ar-h),9.95(s,1h,-conh),3.90-3.91(d,1h,-cochco-),3.56-3.65(m,1h,-ch),2.70-2.72(d,2h,-coch2).
实施例5
称取0.0013mol巴比妥酸和0.0012molnaoh于干燥的研钵中,迅速将其混合均匀,再加入0.001mol1-吩噻嗪基-3-(4-二甲氨基苯基)-丙烯酮,研磨36min。研磨过程中,混合物会逐渐变黏,继续研磨直至物质不再发生变化,使用tlc跟踪监测反应进程,所用的展开剂为体积比为1:5的乙酸乙酯和石油醚的混合溶剂,待反应结束后,用纯净水对产物进行多次水洗抽滤,直到滤液的ph呈中性,然后将滤饼自然风干,得灰色粉末,即为1-吩噻嗪基-3-(4-二甲氨基苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。产率为86.1%,m.p.105℃~108℃。
ir(kbr)ν(cm-1):3057(=c-h,ar-h),3326(-nh-),2972,1380(-ch3),2922,2850,1438(-ch2),1709,1662(c=o),1593,1463(ar),743(苯环上邻二取代),816(苯环上对位取代).
1hnmr:6.64-7.12(m,12h,ar-h),9.86(s,1h,-conh),3.94-3.95(d,1h,-cochco-),3.67-3.72(m,1h,-ch),2.68-2.70(d,2h,-coch2),3.11(s,3h,-nch3).
实施例6
称取0.0015mol巴比妥酸和0.0011mol无水k2co3于干燥的研钵中,迅速将其混合均匀,再加入0.001mol1-吩噻嗪基-3-(4-甲氧基苯基)-丙烯酮,研磨32min。研磨过程中,混合物会逐渐变黏,继续研磨直至物质不再发生变化,使用tlc跟踪监测反应进程,所用的展开剂为体积比为1:5的乙酸乙酯和石油醚的混合溶剂,待反应结束后,用纯净水对产物进行多次水洗抽滤,直到滤液的ph呈中性,然后将滤饼自然风干,得紫色粉末,即为1-吩噻嗪基-3-(4-甲氧基苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。产率为86.35,m.p.114℃~118℃。
ir(kbr)ν(cm-1):3054(=c-h,ar-h),3315(-nh-),2976,1385(-ch3),2920,2847,1442(-ch2),1712,1668(c=o),1593,1462(ar),741(苯环上邻二取代),817(苯环上对位取代).
1hnmr:6.63-7.12(m,12h,ar-h),9.92(s,1h,-conh),3.92-3.93(d,1h,-cochco-),3.65-3.68(m,1h,-ch),2.58-2.60(d,2h,-coch2),4.15(s,3h,-och3).
实施例7
称取0.0014mol巴比妥酸和0.0015mol无水k2co3于干燥的研钵中,迅速将其混合均匀,再加入0.001mol1-吩噻嗪基-3-(4-甲基苯基)-丙烯酮,研磨38min。研磨过程中,混合物会逐渐变黏,继续研磨直至物质不再发生变化,使用tlc跟踪监测反应进程,所用的展开剂为体积比为1:5的乙酸乙酯和石油醚的混合溶剂,待反应结束后,用纯净水对产物进行多次水洗抽滤,直到滤液的ph呈中性,然后将滤饼自然风干,得紫色粉末,即为1-吩噻嗪基-3-(4-甲基苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。产率为86.0%,m.p.118℃~120℃。
ir(kbr)ν(cm-1):3058(=c-h,ar-h),3324(-nh-),2971,1378(-ch3),2851,1440(-ch2),1708,1668(c=o),1594,1462(ar),743(苯环上邻二取代),816(苯环上对位取代).
图8是本发明实施例7制备的1-吩噻嗪基-3-(4-甲基苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮的1hnmr图谱,由图7可知:1hnmr:6.58-7.01(m,12h,ar-h),10.42(s,1h,-conh),3.87-3.90(d,1h,-cochco-),3.59-3.67(m,1h,-ch),2.75-2.77(d,2h,-coch2),2.45(s,3h,-ch3).
实施例8
称取0.0011mol巴比妥酸和0.0013molnaoh于干燥的研钵中,迅速将其混合均匀,再加入0.001mol1-吩噻嗪基-3-(4-硝基苯基)-丙烯酮,研磨31min。研磨过程中,混合物会逐渐变黏,继续研磨直至物质不再发生变化,使用tlc跟踪监测反应进程,所用的展开剂为体积比为1:5的乙酸乙酯和石油醚的混合溶剂,待反应结束后,用纯净水对产物进行多次水洗抽滤,直到滤液的ph呈中性,然后将滤饼自然风干,得紫色粉末,即为1-吩噻嗪基-3-(4-硝基苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。产率为88.2%,m.p.117℃~120℃。
ir(kbr)ν(cm-1):3057(=c-h,ar-h),3325(-nh-),2922,2847,1439(-ch2),1707,1656(c=o),1593,1463(ar),743(苯环上邻二取代),817(苯环上对位取代),1494,1357(-no2).
1hnmr:6.79-8.00(m,12h,ar-h),10.12(s,1h,-conh),3.94-3.96(d,1h,-cochco-),3.70-3.76(m,1h,-ch),2.65-2.67(d,2h,-coch2).
实施例9
称取0.0015mol巴比妥酸和0.0015mol无水k2co3于干燥的研钵中,迅速将其混合均匀,再加入0.001mol1-吩噻嗪基-3-(2-噻吩基)-丙烯酮,研磨39min。研磨过程中,混合物会逐渐变黏,继续研磨直至物质不再发生变化,使用tlc跟踪监测反应进程,所用的展开剂为体积比为1:5的乙酸乙酯和石油醚的混合溶剂,待反应结束后,用纯净水对产物进行多次水洗抽滤,直到滤液的ph呈中性,然后将滤饼自然风干,得紫色粉末,即为1-吩噻嗪基-3-(2-噻吩基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。产率为85.8%,m.p.113℃~116℃。
图5是本发明实施例9制备的1-吩噻嗪基-3-(2-噻吩基)-3-(2,4,6-嘧啶三酮基)-1-丙酮的红外图谱,由图5可知:3058cm-1处为ar-h的振动吸收峰,在3322cm-1处为-nh-的吸收峰,在2922cm-1,2848cm-1,1438cm-1处为-ch2的吸收峰,在1708cm-1,1668cm-1处为c=o的特征吸收峰,1573cm-1和1464cm-1处的峰为苯环的呼吸振动吸收峰,743cm-1为苯环上的邻二取代的吸收峰。综上所述,可以证实合成得到了1-吩噻嗪基-3-(2-噻吩基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。
1hnmr:6.78-6.99(m,8h,ar-h),10.53(s,1h,-conh),3.93-3.95(d,1h,-cochco-),3.67-3.72(m,1h,-ch),2.60-2.62(d,2h,-coch2),6.49-6.65(d,1h,thy-h),6.86-6.88(m,1h,thy-h),7.01-7.03(d,1h,thy-h).
实施例10
称取0.0015mol巴比妥酸和0.0013molnaoh于干燥的研钵中,迅速将其混合均匀,再加入0.001mol1-吩噻嗪基-3-(2-吡啶基)-丙烯酮,研磨30min。研磨过程中,混合物会逐渐变黏,继续研磨直至物质不再发生变化,使用tlc跟踪监测反应进程,所用的展开剂为体积比为1:5的乙酸乙酯和石油醚的混合溶剂,待反应结束后,用纯净水对产物进行多次水洗抽滤,直到滤液的ph呈中性,然后将滤饼自然风干,得灰色粉末,即为1-吩噻嗪基-3-(2-吡啶基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。产率为87.6%,m.p.112℃~115℃。
图6是本发明实施例10制备的1-吩噻嗪基-3-(2-吡啶基)-3-(2,4,6-嘧啶三酮基)-1-丙酮的红外图谱,由图6可知:3058cm-1处为ar-h的振动吸收峰,在3321cm-1处为-nh-的吸收峰,在2929cm-1,2854cm-1,1440cm-1处为-ch2的吸收峰,在1706cm-1,1656cm-1处为c=o的特征吸收峰,1581cm-1和1464cm-1处的峰为苯环的呼吸振动吸收峰,744cm-1为苯环上的邻二取代的吸收峰。综上所述,可以证实合成得到了1-吩噻嗪基-3-(2-吡啶基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。
1hnmr:6.63-7.11(m,8h,ar-h),10.29(s,1h,-conh),3.96-3.98(d,1h,-cochco-),3.70-3.74(m,1h,-ch),2.51-2.58(d,2h,-coch2),7.23-7.25(d,1h,py-h),7.93-8.00(m,1h,py-h),8.77-8.79(d,1h,py-h).
实施例11
称取0.0014mol巴比妥酸和0.0014molnaoh于干燥的研钵中,迅速将其混合均匀,再加入0.001mol1-吩噻嗪基-3-(2-呋喃基)-丙烯酮,研磨40min。研磨过程中,混合物会逐渐变黏,继续研磨直至物质不再发生变化,使用tlc跟踪监测反应进程,所用的展开剂为体积比为1:5的乙酸乙酯和石油醚的混合溶剂,待反应结束后,用纯净水对产物进行多次水洗抽滤,直到滤液的ph呈中性,然后将滤饼自然风干,得灰色粉末,即为1-吩噻嗪基-3-(2-呋喃基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。产率为85.7%,m.p.110℃~114℃。
ir(kbr)ν(cm-1):3058(=c-h,ar-h),3318(-nh-),2922,2849,1438(-ch2),1716,1664(c=o),1592,1463(ar),743(苯环上邻二取代),1040,1126(c-o-c).
1hnmr:6.65-7.03(m,8h,ar-h),10.08(s,1h,-conh),3.91-3.93(d,1h,-cochco-),3.64-3.69(m,1h,-ch),2.55-2.57(d,2h,-coch2),6.03-6.06(d,1h,fu-h),6.29-6.36(m,1h,fu-h),7.23-7.25(d,1h,fu-h).
实施例12
称取0.001mol巴比妥酸和0.001molnaoh于干燥的研钵中,迅速将其混合均匀,再加入0.001mol1-吩噻嗪基-3-(3-硝基苯基)-丙烯酮,研磨35min。研磨过程中,混合物会逐渐变黏,继续研磨直至物质不再发生变化,使用tlc跟踪监测反应进程,所用的展开剂为体积比为1:5的乙酸乙酯和石油醚的混合溶剂,待反应结束后,用纯净水对产物进行多次水洗抽滤,直到滤液的ph呈中性,然后将滤饼自然风干,得紫色粉末,即为1-吩噻嗪基-3-(3-硝基苯基)-3-(2,4,6-嘧啶三酮基)-1-丙酮。产率为89.0%,m.p.106℃~108℃。
ir(kbr)ν(cm-1):3054(=c-h,ar-h),3331(-nh-),2921,2850,1440(-ch2),1712,1667(c=o),1594,1462(ar),744(苯环上邻二取代),1515,1357(-no2).
1hnmr:6.64-8.02(m,12h,ar-h),10.02(s,1h,-conh),3.88-3.90(d,1h,-cochco-),3.70-3.78(m,1h,-ch),2.69-2.71(d,2h,-coch2).
除上述实施例外,原料1-吩噻嗪基-3-芳基-丙烯酮还可采用以下物质,其中ar为邻硝基苯基、羟基苯基、苯乙烯基等;在此不一一列举。
另外,本发明中的1-吩噻嗪基-3-芳基-丙烯酮可采用以下方法制备:
向250ml干燥的三口烧瓶中添加10-乙酰基吩噻嗪(1.21g,0.005mol)、naoh颗粒(0.24g,0.006mol)和无水甲醇15ml,室温下搅拌10min,滴加芳香醛(0.006mol)的无水甲醇溶液10ml,回流反应30min(tlc监测所述反应进程)。反应结束后冷却至室温,将所得反应液于40ml冰水中析出全部固体,抽滤,滤饼用冰水洗涤,自然风干,再用无水甲醇对其进行重结晶,即得到得1-吩噻嗪基-3-芳基-丙烯酮。其反应方程式如下:
其中ar为苯基、对氯苯基、对溴苯基、对氟苯基、甲基苯基、甲氧基苯基、对硝基苯基、间硝基苯基、邻硝基苯基、二甲氨基苯基、羟基苯基、苯乙烯基、呋喃基、噻吩基或吡啶基。
本发明采用固相研磨法,由巴比妥酸和1-吩噻嗪基-3-芳基-丙烯酮反应,合成了一系列未见文献报道的新的michael加成产物,即1-吩噻嗪基-3-芳基-3-(2,4,6-嘧啶三酮基)-1-丙酮,并探索了其制备方法,同时优化了实验条件,缩短了反应时间,为绿色合成做出了贡献。本发明提供的1-吩噻嗪基-3-芳基-3-(2,4,6-嘧啶三酮基)-1-丙酮是一系列全新的含有吩噻嗪基的酮类衍生物,具有杀菌、抑菌等功效,在抗菌、抗病毒等领域有很好的应用前景。
以上所述,仅是本发明的较佳实施例,并非对本发明作任何限制,凡是根据本发明技术实质对以上实施例所作的任何简单修改、变更以及等效结构变换,均仍属于本发明技术方案的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!