一种多取代吡啶的制备方法与流程
本发明涉及一种多取代吡啶的制备方法,属于精细化工产品的制备领域。
背景技术:
吡啶是一类重要的杂环化合物,不但广泛分布于自然界中,而且在药物化学,合成化学,以及材料化学中都有广泛的应用。例如,含吡啶结构的维生素B3(烟酸)和维生素B6(吡哆素)是人和动物内所必须的维生素,是多种辅酶的重要组成部分,用于参与体内的重要代谢活动。NADP也是生物体内一类重要的含吡啶结构的辅酶,在呼吸作用中参与氧化还原过程提供能量。除此之外,在已上市的畅销药物(如吡格列酮,雷贝拉唑,伊马替尼等),pH值荧光指示材料【J.Am.Chem.Soc.2009,131,3016】,以及手型多齿配体【Org.Lett.2007,9,3933】中,均可发现多取代吡啶的结构。因此,由于其在自然界和工业中的重要性,人们希望找到更多绿色环保、合成简便的方法制备该类杂环化合物。
合成吡啶最常用的方法是使用胺与1,5-二羰基化合物缩合,再通过氧化作用将缩合产物二氢吡啶氧化形成多取代吡啶。而通常使用的氧化剂为硝酸【Liebigs Ann.Chem.1882,215,1】,1,4-二苯醌【Eur.J.Org.Chem.2001,2115】,或者单质碘【Synthesis,2000,1532.】,当然使用氧气或者空气作为氧化剂也有相关文献报道,然而通常都需要200度以上的高温【J.Org.Chem.2002,2197】。由于这些氧化过程需要消耗大量的氧化剂以及高温反应不易操作的缺陷,因此能够寻求一些廉价易得、操作简便、绿色环保的氧化剂完成1,5-二羰基化合物到吡啶的转化十分必要。
二甲基亚砜(DMSO)既是一种常见的有机溶剂,又是一类绿色环保的氧化剂。例如经典的氧化反应Swern氧化,就是利用二甲基亚砜的氧化性将伯醇氧化成醛。
技术实现要素:
本发明旨在提供一种多取代吡啶的制备方法,反应条件温和,操作过程简单。
本发明提供了一种多取代吡啶的制备方法,使用1,5-二羰基化合物和醋酸铵为原料,不需要外加任何氧化剂,以二甲基亚砜为溶剂和氧化剂,加热至85-95℃得到1,2,3,5-四取代的吡啶产品;收率可达70-87%;所使用的1,5-二羰基化合物结构如下:
其中R1和R2代表具有不同取代基的苯环;R3代表酯基;R4代表甲基或有不同取代基的苯环。
本发明关于取代基和苯环的定义均为本领域普通技术人员所熟知,在此及下文不再做详细的说明。
本发明的反应方程式如下:
上述多取代吡啶的制备方法,具体操作步骤如下:
(1)将1,5-二羰基化合物溶解在DMSO中,搅拌使其溶解,称取醋酸铵固体一次性加入上述DMSO溶液中,将混合液升温至85-95℃,搅拌8-12小时;
(2)通过薄层层析板检查反应直至原料1,5-二羰基化合物反应完全,反应混合液降至室温用乙酸乙酯稀释,并用去离子水洗涤乙酸乙酯层3~5次;乙酸乙酯层有机相减压蒸干,浓缩物用二氯甲烷重结晶,制得1,2,3,5-四取代的吡啶产品。
上述制备方法中,底物1,5-二羰基化合物与醋酸铵的摩尔比为1:(3-10);每摩尔1,5-二羰基化合物所使用的溶剂DMSO的体积为5-10升。
上述原料配比优选为底物1,5-二羰基化合物与醋酸铵的摩尔比为1:5;每摩尔1,5-二羰基化合物使用溶剂DMSO的体积为5升。
上述制备方法中,反应最佳条件为:反应温度为90℃,反应时间为8小时。
本发明为1,2,3,5-四取代吡啶特别是3位具有酯基的取代吡啶提供了一种简便、绿色、高效的合成方法。
本发明的反应机理如下所述:醋酸铵在加热的条件下释放出氨气分子与含有酯基的1,5-二羰基化合物缩合形成烯胺,烯胺发生分子内缩合并通过异构化形成二氢吡啶;在DMSO的氧化作用下,二氢吡啶被氧化,并最终脱去水分子形成吡啶。
本发明的有益效果:
本方法使用二甲基亚砜(DMSO)为溶剂和氧化剂,在加热的条件下可以完成1,5-二羰基化合物与醋酸铵反应生成1,2,3,5-四取代吡啶。反应体系简单,避免了额外添加氧化剂以及高温强热的条件,尤其针对于3位有酯基取代的吡啶结构有较好的适用性。
附图说明
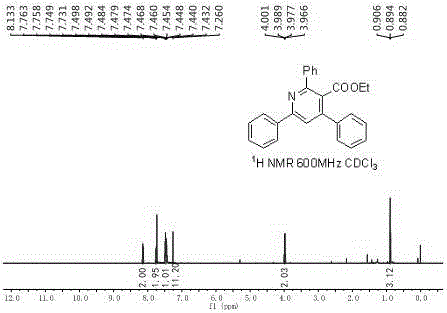
图1为实施例1产品的核磁共振氢谱图。
图2为实施例2产品的核磁共振氢谱图。
图3为实施例3产品的核磁共振氢谱图。
图4为实施例4产品的核磁共振氢谱图。
图5为实施例5产品的核磁共振氢谱图。
图6为实施例6产品的核磁共振氢谱图。
图7为实施例7产品的核磁共振氢谱图。
具体实施方式
下面通过实施例来进一步说明本发明,但不局限于以下实施例。
实施例1:
在500mL的圆底烧瓶中,加入20g(50mmol)2-苯甲酰基-5-氧杂-3,5-二苯基戊酸乙酯,搅拌使其溶解于250mL二甲基亚砜中。将20g(250mmol)醋酸铵固体加入到上述混合液中,室温搅拌使其溶解。待醋酸铵完全溶解后,将反应体系升至90℃搅拌8小时,并用薄层层析板检测反应进度。
反应结束,停止搅拌并将反应液降至室温,加入500mL乙酸乙酯稀释反应液,并加入蒸馏水洗涤乙酸乙酯层3-5次,每次用水250mL。洗涤结束加入干燥剂干燥有机相,浓缩后用二氯甲烷重结晶得到白色固体15.2g,产率80%,熔点86-88℃。
所得产物经核磁鉴定为目标产品。
如图1所示,1H NMR(CDCl3,600MHz):δ8.15-8.12(m,2H),7.77-7.74(m,2H),7.73(s,1H),7.51-7.43(m,11H),3.98(q,2H,J=7.2Hz),0.89(t,3H,J=7.2Hz)。
实施例2
在500mL的圆底烧瓶中,加入21g(50mmol)2-苯甲酰基-5-氧杂-3-苯基-5-对甲苯基戊酸乙酯,搅拌使其溶解于350mL二甲基亚砜中。将12g(150mmol)醋酸铵固体加入到上述混合液中,室温搅拌使其溶解。待醋酸铵完全溶解后,将反应体系升至85℃搅拌10小时,并用薄层层析板检测反应进度。
反应结束,停止搅拌并将反应液降至室温,加入500mL乙酸乙酯稀释反应液,并加入蒸馏水洗涤乙酸乙酯层3-5次,每次用水250mL。洗涤结束加入干燥剂干燥有机相,浓缩后用二氯甲烷重结晶得到白色固体14.3g,产率73%,熔点77-79℃。
所得产物经核磁鉴定为目标产品。
如图2所示,1H NMR(CDCl3,400MHz):δ8.05(d,2H,J=8.0Hz),7.75(d,2H,J=6.4Hz),7.70(s,1H),7.52-7.41(m,8H),7.30(d,2H,J=7.6Hz),3.98(q,2H,J=7.2Hz),2.42(s,3H,),0.89(q,3H,J=7.2Hz)。
实施例3
在1L的圆底烧瓶中,加入22g(50mmol)2-苯甲酰基-5-氧杂-3-苯基-5-对硝基苯基戊酸乙酯,搅拌使其溶解于500mL二甲基亚砜中。将39g(500mmol)醋酸铵固体加入到上述混合液中,室温搅拌使其溶解。待醋酸铵完全溶解后,将反应体系升至95℃搅拌12小时,并用薄层层析板检测反应进度。
反应结束,停止搅拌并将反应液降至室温,加入1L乙酸乙酯稀释反应液,并加入蒸馏水洗涤乙酸乙酯层3-5次,每次用水250mL。洗涤结束加入干燥剂干燥有机相,浓缩后用二氯甲烷重结晶得到白色固体15.9g,产率75%,熔点151-153℃。
所得产物经核磁鉴定为目标产品。
如图3所示,1H NMR(CDCl3,400MHz):δ8.36-8.30(m,4H),7.80(s,1H),7.76-7.72(m,2H),7.51-7.45(m,8H),4.00(q,2H,J=7.2Hz),0.90(t,3H,J=7.2Hz)。
实施例4
在500mL的圆底烧瓶中,加入21.5g(50mmol)2-苯甲酰基-5-氧杂-3-对甲氧基苯基-5-苯基戊酸乙酯,搅拌使其溶解于400mL二甲基亚砜中。将31g(400mmol)醋酸铵固体加入到上述混合液中,室温搅拌使其溶解。待醋酸铵完全溶解后,将反应体系升至85℃搅拌10小时,并用薄层层析板检测反应进度。
反应结束,停止搅拌并将反应液降至室温,加入500mL乙酸乙酯稀释反应液,并加入蒸馏水洗涤乙酸乙酯层3-5次,每次用水250mL。洗涤结束加入干燥剂干燥有机相,浓缩后用二氯甲烷重结晶得到白色固体14.3g,产率70%,熔点148-150℃。
所得产物经核磁鉴定为目标产品。
如图4所示,1H NMR(CDCl3,400MHz):δ8.04(d,2H,J=8.0Hz),7.77-7.72(m,2H),7.70(s,1H),7.50-7.41(m,8H),7.29(d,2H,J=8.0Hz),3.98(q,2H,J=7.2Hz),2.42(s,3H),0.89(t,3H,J=7.2Hz)。
实施例5
在500mL的圆底烧瓶中,加入21.7g(50mmol)2-苯甲酰基-5-氧杂-3-对氯苯基-5-苯基戊酸乙酯,搅拌使其溶解于300mL二甲基亚砜中。将20g(250mmol)醋酸铵固体加入到上述混合液中,室温搅拌使其溶解。待醋酸铵完全溶解后,将反应体系升至90℃搅拌10小时,并用薄层层析板检测反应进度。
反应结束,停止搅拌并将反应液降至室温,加入500mL乙酸乙酯稀释反应液,并加入蒸馏水洗涤乙酸乙酯层3-5次,每次用水250mL。洗涤结束加入干燥剂干燥有机相,浓缩后用二氯甲烷重结晶得到白色固体18.0g,产率87%,熔点122-124℃。
所得产物经核磁鉴定为目标产品。
如图5所示,1H NMR(CDCl3,600MHz):δ8.14-8.10(m,2H),7.76-7.72(m,2H),7.68(s,1H),7.51-7.40(m,10H),3.99(q,2H,J=7.2Hz),0.92(t,3H,J=7.2Hz)。
实施例6
在500mL的圆底烧瓶中,加入16.9g(50mmol)2-乙酰基-5-氧杂-3,5-二苯基戊酸乙酯,搅拌使其溶解于250mL二甲基亚砜中。将16g(200mmol)醋酸铵固体加入到上述混合液中,室温搅拌使其溶解。待醋酸铵完全溶解后,将反应体系升至90℃搅拌8小时,并用薄层层析板检测反应进度。
反应结束,停止搅拌并将反应液降至室温,加入500mL乙酸乙酯稀释反应液,并加入蒸馏水洗涤乙酸乙酯层3-5次,每次用水250mL。洗涤结束加入干燥剂干燥有机相,浓缩后用二氯甲烷重结晶得到无色液体12.2g,产率77%。
如图6所示,1H NMR(CDCl3,100MHz):δ8.08-8.01(m,2H),7.58(s,1H),7.53-7.39(m,8H),4.13(q,2H,J=7.2Hz),2.74(s,3H),1.01(t,3H,J=7.2Hz)。
实施例7
在500mL的圆底烧瓶中,加入20.0g(50mmol)2-乙酰基-5-氧杂-3,5-二苯基戊酸苄酯,搅拌使其溶解于300mL二甲基亚砜中。将23g(300mmol)醋酸铵固体加入到上述混合液中,室温搅拌使其溶解。待醋酸铵完全溶解后,将反应体系升至90℃搅拌9小时,并用薄层层析板检测反应进度。
反应结束,停止搅拌并将反应液降至室温,加入500mL乙酸乙酯稀释反应液,并加入蒸馏水洗涤乙酸乙酯层3-5次,每次用水250mL。洗涤结束加入干燥剂干燥有机相,浓缩后用二氯甲烷重结晶得到无色液体15.0g,产率79%。
如图7所示,1H NMR(CDCl3,400MHz):δ8.01(d,2H,J=7.2Hz),7.56(s,1H),7.50-7.36(m,8H),7.25-7.21(m,3H),7.02-6.96(m,2H),5.07(s,2H),2.70(s,2H)。
- 还没有人留言评论。精彩留言会获得点赞!