一种2‑(芘‑1‑基)‑1,8‑萘啶及其合成方法与流程
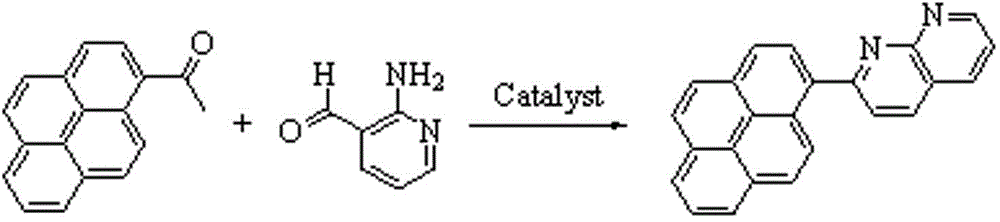
本发明属于有机物合成技术领域,具体涉及一种2-(芘-1-基)-1,8-萘啶及其合成方法。
背景技术:
甲基芳基酮与芳香醛一般在碱性条件下能够发生缩合反应,生成查尔酮;选用邻位带有氨基的3-吡啶甲醛与1-乙酰基芘反应,在一定的温度和时间条件下反应生成了萘啶衍生物,结果是芘和萘啶两个芳香体系通过碳碳单键的建立而联在一起,形成更大的芳香共轭体系;这种更大芳香共轭体系的化合物更易于在强激光作用下产生非线性光学效应。
技术实现要素:
本发明为了解决现有技术中1-乙酰芘和2-氨基吡啶-3-甲醛在不同反应条件时产物不同的问题,提供一种2-(芘-1-基)-1,8-萘啶及其合成方法。
本发明采用如下技术方案:
一种2-(芘-1-基)-1,8-萘啶,其结构式如式Ⅰ:
式Ⅰ。
所述2-(芘-1-基)-1,8-萘啶的合成方法,包括如下步骤:
在带有回流装置的250mL三口圆底烧瓶中依次加入0.61g1-乙酰芘、0.43g2-氨基吡啶-3-甲醛和90mL无水乙醇,待固体完全溶解后加入催化剂,60-80℃磁力搅拌反应3-5h,反应期间TLC跟踪反应,反应完成后,停止搅拌,冷却,抽滤,用无水乙醇洗涤滤饼至不再有反应物,真空干燥,得黄色粉末固体0.65-0.67g,产率79-81%;熔点195~196℃。
所述催化剂为20%NaOH溶液,添加量为1-5mL。
所述TLC跟踪反应的展开剂为V乙酸乙酯:V石油醚= 1:3。
为说明本发明的2-(芘-1-基)-1,8-萘啶的结构,对本发明化合物分别采用IR、1H NMR、13C NMR、LCMS进行了结构表征;并测定了紫外光谱、荧光光谱和光学非线性吸收性能。
将本发明化合物配制成浓度为1×10-5mol/L的二氯甲烷溶液,测定它们的紫外吸收光谱;
将本发明化合物配制成浓度为1×10-6 mol/L的二氯甲烷溶液,测定它们的荧光发射光谱;
测样条件:(1)色谱条件:两通进样,流 动 相:A相-水溶液;B相-甲醇,A:B=30:70,流速:0.4 mL/min,进样体积:5 μL;(2)质谱条件:离子源: ESI,负离子扫描,雾化气: 氮气 3.0 L/min,干燥气:氮气 15 L/min,碰撞气:氩气,脱溶剂管温度:400℃,加热模块温度250℃,扫描模式:Q3Scan,驻留时间:100 ms。在上述条件下测定本发明化合物的质谱。
非线性光学吸收测定:采用Z-扫描技术测定,激光器:Nd: YAG (激光脉冲21 ps,波长532 nm,实验重复频率10 Hz),数值拟合获得非线性光学吸收系数β= 9.0×10-14 m/W。
本发明化合物结构表征如下:
I.R.νmax/cm-1(KBr):3039.91,1599.04,1489.10,1425.44;
1H NMR(600MHz,DMSO-d),δ/ppm:9.205-9.199(t,1H,J=1.8),8.733-8.719(d,1H,J=8.4),8.649-8.632(d,1H,J=10.2Hz),8.584-8.569(d,1H,J=9Hz),8.497-8.483(d,1H,J=8.4Hz),8.406-8.358(m,3H),8.317(s,2H),8.272-8.257(d,1H,J=9Hz),8.163-8.139(t,2H,J=7.2Hz),7.761-7,740(q,1H,J=4.2Hz);
13C NMR(150MHz,DMSO-d),δ/ppm:162.40,155.88,154.61,138.89,138.07,135.32,131.79,131.38,130.80,128.77,128.66,128.59,127.86,127.07,126.30,125.91,125.46,125.17,125.11,124.64,124.35,123.06,121.82;
LC-MS(ESI) m/z:found:331.10;Calcd. for C24H15N2([M+H]+):331.38。
红外光谱显示了2-(芘-1-基)-1,8-萘啶的各官能团特征吸收峰;核磁共振氢谱显示氢原子的种类与个数与2-(芘-1-基)-1,8-萘啶所含相吻合,碳谱显示碳原子种类与分子中所含相一致;LCMS(ESI)显示实测值与计算值相差极小,相对误差0.069‰;综合各谱分析认为,合成的2-(芘-1-基)-1,8-萘啶结构正确。
本发明的有益效果如下:
1.在催化剂作用下,得到所需产物2-(芘-1-基)-1,8-萘啶;
2.本发明催化剂选择性强,方法简便,产率较高。
附图说明
图1为本发明化合物合成原理图;
图2为本发明化合物IR光谱图;
图3为本发明化合物1H NMR图;
图4为 图3局部放大图;
图5为本发明化合物13C NMR图;
图6为图5局部放大图;
图7为本发明化合物的LCMS图;
图8为本发明化合物的UV图;
图9为本发明化合物的荧光发射谱图;
图10为本发明化合物的光学非线性吸收图。(激光波长532nm在3.95uj能量下(样品/DMSO)的开孔Z扫描数据。点表示实验数据,线是理论拟合结果。)。
具体实施方式
一种2-(芘-1-基)-1,8-萘啶,其结构式如式Ⅰ:
式Ⅰ。
实施例1,所述2-(芘-1-基)-1,8-萘啶的合成方法,包括如下步骤:
在带有回流装置的250mL三口圆底烧瓶中依次加入0.61g 1-乙酰基芘、0.43g2-氨基吡啶-3-甲醛和90mL无水乙醇,待固体完全溶解后加入催化剂1mL 20%NaOH溶液,60℃磁力搅拌反应3h,反应期间TLC跟踪反应(展开剂为V乙酸乙酯:V石油醚= 1:3),反应完成后,停止搅拌,冷却,抽滤,用无水乙醇洗涤滤饼至不再有反应物,真空干燥,得黄色粉末固体0.65g,产率79%;熔点195℃。
实施例2,所述2-(芘-1-基)-1,8-萘啶的合成方法,包括如下步骤:
在带有回流装置的250mL三口圆底烧瓶中依次加入0.61g 1-乙酰基芘、0.43g2-氨基吡啶-3-甲醛和90mL无水乙醇,待固体完全溶解后加入催化剂3mL 20%NaOH溶液,70℃磁力搅拌反应4h,反应期间TLC跟踪反应(展开剂为V乙酸乙酯:V石油醚= 1:3),反应完成后,停止搅拌,冷却,抽滤,用无水乙醇洗涤滤饼至不再有反应物,真空干燥,得黄色粉末固体0.66g,产率80%;熔点195.5℃。
实施例3,所述2-(芘-1-基)-1,8-萘啶的合成方法,包括如下步骤:
在带有回流装置的250mL三口圆底烧瓶中依次加入0.61g 1-乙酰基芘、0.43g2-氨基吡啶-3-甲醛和90mL无水乙醇,待固体完全溶解后加入催化剂5mL 20%NaOH溶液,80℃磁力搅拌反应5h,反应期间TLC跟踪反应(展开剂为V乙酸乙酯:V石油醚= 1:3),反应完成后,停止搅拌,冷却,抽滤,用无水乙醇洗涤滤饼至不再有反应物,真空干燥,得黄色粉末固体0.67g,产率81%;熔点196℃。
- 还没有人留言评论。精彩留言会获得点赞!