体液循环DNA定量检测方法及试剂盒与流程
本发明属于生物样品检测领域,涉及体液循环DNA定量检测方法及试剂盒。
背景技术:
循环DNA(circulating DNA)又称为无细胞状态DNA(cell-free DNA),存在于体液,如血液(血浆或血清)、尿液、脑脊液以及滑膜液的细胞外DNA。体液循环DNA的定量检测结果受到多种因素影响,会产生较大偏差,其中在核酸提取和长期保存过程中DNA的丢失和降解是其中两个最主要的影响因素。
由于体液样本中循环DNA含量较低,因此需要采用基因扩增手段进行定量分析。基因扩增检测需要对核酸进行提取,由于提取过程中核酸的丢失,其定量结果无法反映体液循环DNA的真实水平;另一方面,各种核酸提取方法的提取效率存在很大差异,且核酸提取的重复性差,会在很大程度上影响定量检测结果的一致性。同时,无论是原始或纯化后的DNA样本,在长期保存过程中存在不同程度的降解,DNA年降解率甚至高达30%,这种变化对于定量检测结果的准确性和最终结论会产生巨大的影响。正因为如此,体液循环DNA定量检测分析一直都没有准确稳定的标准化方法。
传统的体液循环DNA定量方法包括荧光定量PCR外标法和PicoGreen荧光法。荧光定量PCR外标法是针对人看家基因的单重荧光定量PCR,通过已知浓度的外部标准品建立标准曲线,对待测样本进行定量分析,由于标准品和待测样本分别在不同的试管中进行操作,其管间差异较大,影响定量结果的精密度和稳定性。PicoGreen荧光法采用PicoGreen荧光染料与双链DNA非特异性结合,结合后荧光强度显著增加,通过已知浓度的外部标准品建立标准曲线,从而对待测样本进行定量分析,由于是直接检测而未对DNA进行扩增,该方法检测灵敏度较荧光定量PCR法低,且存在较大的管间差异,影响定量结果的精密度和稳定性。为了解决上述问题,发明人前期设计了血浆循环DNA内标法定量检测,详见CN1811387,虽然较PCR外标法和PicoGreen荧光法有一定程度的改进,然而由于内标序列与引物序列设计的问题,造成内标与目的基因扩增目的片段长度不同,导致两者在双重荧光PCR反应过程中的扩增效率不一致,对其定量检测结果仍然存在显著影响。
技术实现要素:
本发明的目的是针对现有技术的上述不足,提供一种体液循环DNA定量检测方法。
本发明的另一目的是提供一种体液循环DNA定量检测试剂盒。
一种非疾病诊断与治疗用途的体液循环DNA定量检测方法,包括:
人工合成带有“A”粘性末端、长度为45bp的线性双链DNA片段,线性双链DNA片段中有25bp序列与β-actin基因相同,该线性双链DNA片段与T-Vector pMD20质粒载体重组后,得到长度为2781bp的环状重组质粒DNA,经限制性内切酶Sma I酶切为线性双链DNA和紫外分光光度法定量后以一定量加入到待测样本中作为内标(图1);提取待测样本的总DNA,同时包含待测循环DNA和内标,采用双重实时荧光定量PCR法同时扩增循环DNA中的人看家基因β-actin和内标,按照内标的浓度计算被测体液中β-actin基因浓度,以此得出循环DNA含量;其中,双重实时荧光定量PCR反应体系中包含:共用下游引物、分别针对内标和β-actin基因的上游引物、以及分别标记JOE和FAM荧光基团的两个Taqman水解探针,JOE标记的探针针对β-actin基因,FAM标记的探针针对内标,同步进行基因扩增定量检测;所述的线性双链DNA片段序列如SEQ ID NO.1所示;
在双重实时荧光定量PCR扩增内标时,上游引物如SEQ ID NO.2所示,下游引物如SEQ ID NO.3所示,Taqman水解探针如SEQ ID NO.4所示,5’端标记荧光报告基团FAM;
在双重实时荧光定量PCR扩增β-actin基因时,上游引物如SEQ ID NO.5所示,共用下游引物同内标扩增下游引物如SEQ ID NO.3所示,Taqman水解探针序列如SEQ ID NO.6所示,5’端标记荧光报告基团JOE。
本发明所述的非疾病诊断与治疗用途的体液循环DNA定量检测方法优选:每200ul体液样本中加入浓度为1×104拷贝/ul的内标5ul,内标总拷贝数为5×104,充分混匀后立即检测或可长期-20℃以下冷冻保存。
本发明所述的非疾病诊断与治疗用途的体液循环DNA定量检测方法优选:PCR扩增总反应体系25μl,其中,10×Real Time PCR buffer 2.5μl;dNTPs 400μM;MgCl2 6mM;内标和β-actin基因的上游引物各200nM;共用下游引物700nM;内标和β-actin基因的TaqMan水解探针各100nM;TaKaRa Ex热启动DNA聚合酶1U;循环DNA为2.5μl。
本发明所述的非疾病诊断与治疗用途的循环DNA定量检测方法优选:PCR扩增反应条件:(1)95℃,5min(2)94℃,30s;55℃,30s;72℃,40s,分别在JOE和FAM荧光信号通道上检测一次荧光信号,共40个循环。
本发明所述的非疾病诊断与治疗用途的体液循环DNA定量检测方法优选:①双重实时荧光定量PCR结果采用7500system SDS软件进行分析,分别在JOE和FAM两个通道设定基线和阈值,基线起始点为6或其它不小于5的数,终点为在荧光信号开始升高处再往前2个循环数,阈值设定在低荧光信号指数扩增区,以此确定目的基因β-actin和内标扩增Ct值,以Ctb和Cti表示;②采用LinRegPCR软件,计算各样本的PCR扩增效率,以E表示;③以下述公式计算血浆DNA含量,单位为拷贝/ml,并以1拷贝双链DNA≈3.3×10-3ng计算血浆DNA浓度,单位为ng/ml:
Cb=Ci×(1+E)(Cti-Ctb)
公式中Cb和Ci分别表示血浆中β-actin基因和内标的起始模板浓度,单位为拷贝/ml,E表示PCR扩增效率,Ctb和Cti分别表示β-actin基因和内标的临界循环数。
一种体液循环DNA定量检测试剂盒,其特征在于包含以下组分:
(1)内标:含序列如SEQ ID NO.1所示的线性双链DNA片段的T-Vector pMD20重组线性质粒;
(2)如SEQ ID NO.2所示的内标的上游引物;
(3)如SEQ ID NO.4所示且5’端标记荧光报告基团FAM的内标Taqman水解探针序列;
(4)如SEQ ID NO.5所示的β-actin基因的上游引物;
(5)如SEQ ID NO.6所示且5’端标记荧光报告基团JOE的β-actin基因的Taqman水解探针序列;
(6)如SEQ ID NO.3所示的内标和β-actin基因共用的下游引物。
其中,内标的浓度优选1.0×104拷贝/μl。
本发明试剂盒中还包含10Real Time PCR buffer、dNTPs、MgCl2、TaKaRa Ex热启动DNA聚合酶等组分。
有益效果:
针对现有技术存在的问题,本发明进行了如下改进:①重新设计了插入的双链DNA片段,CN1811387中为避免内标与β-actin基因相互干扰,设计的人工合成DNA片段长度为41bp,且与β-actin基因具有完全不同的序列,与人类基因组也完全不同源;本发明通过大量的实验探索,发现将线性双链DNA片段长度增长至45bp,且其中有25bp序列与β-actin基因相同不仅不会造成内标与β-actin基因相互干扰,并且能够提高扩增效率的一致性;②采用线性质粒载体T-Vector pMD20;③重新设计了共用下游引物序列;④内标和β-actin基因扩增产物长度均为91bp。通过上述措施的联合使用,使得β-actin基因和内标能够在同一反应管中进行效率相同且互不干扰的PCR扩增反应,其扩增信号能够被分别检测,从而显著提高了定量方法的性能,经过与我们前期内标法(即CN1811387中权利要求1请求保护的方法,下同)以及其它2种传统方法(即荧光定量PCR外标法和PicoGreen荧光法,下同)的比较,该体液循环DNA定量检测方法具有更高的精密度、线性范围、准确性和稳定性。
附图说明
图1内标制备流程图
图2克隆载体T-Vector pMD20部分序列及外源片段插入位点
图3循环DNA定量方法的线性范围
图4循环DNA定量方法的回收率试验,图中每一系列从左至右依次为本发明方法、前期内标法、外标法、荧光法。
图5不同提取试剂或DNA聚合酶的循环DNA定量检测结果
图6健康体检者血浆循环DNA定量结果
图7健康体检者尿液循环DNA定量结果
具体实施方式
实施例1
(1)内标的合成
人工合成带有“A”粘性末端的线性双链DNA片段,序列如下:
5’-ACGGGAAGCGGTTGGTGGTGCGGGAAATCGTGCGTGACATTAAGA-3’
3’-ATGCCCTT CGCCAACCACCACGCCC TTT AGCACGCACTGTAATTC-5’
将人工设计的两条互补寡核苷酸分别用Tris-EDTA缓冲液稀释为20μmol/L,等体积混匀,置100℃沸水浴中10min,再放于室温自然冷却得到线性双链DNA片段;将线性双链DNA片段重组至质粒载体T-Vector pMD20中(插入位点见图2),转染至E.coli JM109感受态细胞,体外培养增殖后接种至含有X-Gal、IPTG、Amp的L-琼脂平板培养基上,直接筛选含有重组克隆的白色大菌落,再采用M13测序通用引物RV和M4进行菌落PCR产物电泳分析和测序验证。提取重组质粒DNA并纯化,经限制性内切酶Sma I酶切为线性双链DNA,紫外分光光度法测定线性重组质粒DNA浓度,稀释至1.0×104拷贝/μl,作为定量检测内标。
(2)样本处理和保存
采用两步离心法处理待测体液样本:于室温1600g水平离心10min,小心吸取上清于1.5ml离心管中,再于4℃16000g离心10min,吸取上层无细胞体液样本,每200μl样本中加入浓度为1.0×104拷贝/μl的内标5μl,振荡混匀后-70℃保存待用。
(3)体液循环DNA提取纯化
采用传统酚-氯仿法或商品化核酸提取试剂盒(如柱吸附、磁珠法等),提取体液循环DNA。
(4)双重荧光定量PCR
1)针对β-actin基因的荧光定量PCR引物和探针序列
上游引物(24bp):5’-GGACCTGACTGACTACCTCATGAA-3’(SEQ ID NO.5)
下游引物(25bp):5’-CTTAATGTCACGCACGATTTCCCGC-3’(SEQ ID NO.3)
Taqman探针(22bp):5’-JOE-CACCGAGCGCGGCTACAGCTTC-ECLIPSE-3’(SEQ ID NO.6)
β-actin基因扩增产物序列(91bp):
5’-GGACCTGACTGACTACCTCATGAAGATCCTCACCGAGCGCGGCTACAGCTTCACCACCACGGCCGAGCGGGAAATCGTGCGTGACATTAAG-3’(SEQ ID NO.8)
2)针对内标的荧光定量PCR引物和探针序列
上游引物(24bp):5’-TGCATGCCTGCAGGTCGACTCTAG-3’(SEQ ID NO.2)
下游引物(25bp):5’-CTTAATGTCACGCACGATTTCCCGC-3’(SEQ ID NO.3)
Taqman探针(22bp):5’-FAM-CGCTTCCCGTAATCCATATGAC-ECLIPSE-3’(SEQ ID NO.4)
内标扩增产物序列(91bp):
5’-TGCATGCCTGCAGGTCGACTCTAGAGGATCTACTAGTCATATGGATTACGGGAAGCGGTTGGTGGTGCGGGAAATCGTGCGTGACATTAAG-3’(SEQ ID NO.7)
PCR扩增反应体系:总体系为25μl。其中,10×Real Time PCR buffer 2.5μl;dNTPs 400μM;MgCl2 6mM;上游引物各200nM;共用下游引物700nM;TaqMan水解探针各100nM;TaKaRaEx热启动DNA聚合酶1U;循环DNA为2.5μl。
PCR扩增反应条件:(1)95℃,5min(2)94℃,30s;55℃,30s;72℃,40s,分别在JOE和FAM荧光信号通道上检测一次荧光信号(共40个循环)。
(5)结果分析和计算
①荧光定量PCR结果采用7500system SDS软件进行分析,分别在FAM和JOE两个通道设定基线和阈值。基线起始点为6或其它不小于5的数,终点为在荧光信号开始升高处再往前2个循环数,阈值设定在低荧光信号指数扩增区,以此确定目的基因β-actin和内标扩增Ct值,以Ctb和Cti表示。②采用LinRegPCR软件,计算各样本的PCR扩增效率,以E表示。③以下述公式计算体液循环DNA含量(拷贝/ml),并以1拷贝双链DNA≈3.310-3ng计算体液循环DNA浓度(ng/ml)。
Cb=Ci×(1+E)(Cti-Ctb)
公式中Cb和Ci分别表示体液样本中β-actin基因和内标的起始模板浓度(拷贝/ml),E表示PCR扩增效率,Ctb和Cti分别表示β-actin基因和内标的临界循环数。
实施例2方法学考察
所有样本来自健康体检者(50例),采用EDTA-K2抗凝真空采血管采集肘静脉血(2ml/管),室温静置96小时后,于室温1600g水平离心10min,小心吸取上清于1.5ml离心管中,再于4℃16000g离心10min,吸取上层无细胞血浆,混合后磁珠法提取血浆DNA,紫外分光光度法测定吸光度值并计算DNA浓度,将混合血浆用生理盐水稀释为1000、100、10、1.0和0.1ng/ml不同浓度。分别采用本发明方法、前期内标法、传统外标法和荧光法进行检测比较,方法的具体步骤如下:
本发明:参见“实施例1”。
前期内标法:参见CN1811387中的说明书“具体实施方式”。
外标法:①PCR扩增反应体系:总体系为25μl。其中,10 Real Time PCR buffer 2.5μl;dNTPs400μM;MgCl26mM;上游和下游引物各200nM;TaqMan水解探针100nM;TaKaRa Ex热启动DNA聚合酶1U;血浆DNA模板投入量为2.5μl。②PCR扩增反应条件:95℃,5min预变性,再94℃,30s;55℃,30s;72℃,40s,循环反应40次,每次反应在JOE信号通道上检测一次荧光信号。③结果计算:荧光定量PCR结果采用7500system SDS软件进行分析,在JOE荧光信号通道设定基线和阈值。基线起始点为6或其它不小于5的数,终点为在荧光信号开始升高处再往前2个循环数,阈值设定在低荧光信号指数扩增区,以此确定目的基因β-actin扩增Ct值,以浓度分别0.1、1.0、10、100和1000ng/ml的基因组DNA作为外部标准品,建立定量检测标准曲线,以此计算待测样本的血浆DNA含量。
荧光法:将PicoGreen荧光染料用TE缓冲液(pH=7.0)200倍稀释成工作液,50μl血浆DNA样本与50μl PicoGreen工作液混匀,室温避光孵育5min,以480nm为激发光源波长,在荧光分光光度计中检测520nm波长的荧光信号,并以浓度分别为0.1、1.0、10、100和1000ng/ml的λDNA为外部标准品,建立定量检测标准曲线,以此计算待测样本的血浆DNA含量。
(1)精密度:如表1所示,对于不同浓度的样本,本发明方法的批内和批间CV值均小于前期内标法和传统方法,特别是对于浓度为1.0ng/ml及其以下的样本,本发明方法的检测精密度较其他方法显著提高。
表1血浆循环DNA定量方法的批内和批间CV值
(2)线性范围:如图3所示,本发明方法对0.1-1000ng/ml范围内的循环DNA定量检测的线性相关系数r2达到0.996(图3A),较前期内标法的线性相关性显著提高(图3B);传统外标法和PicoGreen荧光法的线性范围为1-1000ng/ml(r2分别为0.965和0.961)。
(3)准确度:由于经典核酸定量方法检测灵敏度低,对于微量的循环DNA无法检出,缺乏对照方法,因此对于准确度我们采用回收率实验进行评价,结果如图4所示,回收率越接近100%,表明该方法的准确性更高。PCR外标法对1000、100、10和1ng/ml循环DNA的回收率均值分别为73.1%、77.9%、57.3%和48.6%、;PicoGreen荧光法分别为75.2%、74.6%、61.1%和50.7%、,均低于100%,差异有统计学意义(P<0.01)。前期内标法对1000、100和10ng/ml循环DNA的回收率均值分别为99.4%、93.7%和94.1%,与100%的回收率比较,差异无统计学意义(P值均大于0.05);但是对于1ng/ml的样本回收率均值为86.2%,低于100%,差异有统计学意义(P<0.01),表明前期内标法对于浓度为1ng/ml的样本无法准确定量。本发明方法对1000、100、10和1ng/ml循环DNA的回收率均值分别为100.3%、98.7%、102.8%和96.5,与100%的回收率比较,差异无统计学意义(P值均大于0.05),显示了较高的准确性。
(4)稳定性:本发明和传统方法对采用4种不同试剂提取的循环DNA进行定量检测,结果见图5A。由于4种试剂对循环DNA提取效率各不相同,采用传统外标法和荧光法的定量结果各不相同,差异有统计学意义(P<0.05);前期内标法结果虽然有所改善,但仍然存在差异(P<0.05);而本发明方法对采用不同试剂提取的循环DNA定量结果没有差异(P=0.956)。该结果表明,无论采用何种提取方法,虽然存在不同程度的核酸提取丢失,本发明能够通过内标的校准作用,得到稳定可靠的定量检测结果。
本发明和前期内标法以及传统PCR外标法采用4种不同厂家(ABI、MBI、Promega和Takara)的Taq DNA聚合酶进行荧光定量PCR扩增,其循环DNA定量结果见图5B。采用不同Taq DNA聚合酶进行内标法测定的血浆DNA定量结果之间的差异无统计学意义,而前期内标法和外标法的差异有统计学意义(P<0.001)。该结果表明本发明方法不受DNA聚合酶活性的影响,检测结果更加稳定可靠。
实施例3健康体检者血浆样本检测
400例健康体检者,年龄25-60岁,平均年龄43岁,男性200例,女性200例。收集EDTA-K2抗凝外周静脉血,两步离心法分离获得无细胞血浆标本,采用磁珠法试剂盒提取血浆DNA,分别采用本发明方法、前期内标法、传统外标法和荧光法进行定量测定,结果如图6。本发明方法检测结果显著高于前期内标法、传统外标法和荧光法,差异均有统计学意义(P<0.001)。本发明通过内标的内参照作用,消除了核酸提取丢失造成的测量误差,因此其定量结果高于其他方法,且趋势相对集中,并且发现男性血浆DNA水平显著高于女性,而其它方法并未发现此差异。
实施例4健康体检者尿液样本检测
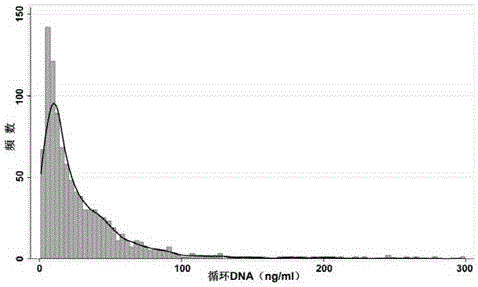
1000例健康体检者,年龄25-60岁,平均年龄42岁,男性500例,女性500例。收集晨尿标本,两步离心法分离获得无细胞尿液标本,采用磁珠法试剂盒提取尿液DNA,采用本发明方法进行定量测定,结果如图7。健康体检者尿液DNA定量范围为1.0~296.4ng/ml(中位值:18.1ng/ml;四分位距:30.5ng/ml),其中84%其尿液DNA在50ng/ml以下,12%在50-100ng/ml之间,4%超过100ng/ml,尿液DNA水平在该1000例健康人群中呈正偏态分布(图7)。不同性别或年龄组之间的尿液DNA含量差异无统计学意义。
<110> 南京麦科林生物医药科技有限公司
<120> 体液循环DNA定量检测方法及试剂盒
<160> 8
<210> 1
<211> 45
<212> DNA
<213> 人工序列
<220>
<223> 线性双链DNA片段正义链序列
<400> 1
acgggaagcg gttggtggtg cgggaaatcg tgcgtgacat taaga 45
<210> 2
<211> 24
<212> DNA
<213> 人工序列
<220>
<223> 针对内标的荧光定量PCR上游引物
<400> 2
tgcatgcctg caggtcgact ctag 24
<210> 3
<211> 25
<212> DNA
<213> 人工序列
<220>
<223> 针对内标的荧光定量PCR下游引物
<400> 3
cttaatgtca cgcacgattt cccgc 25
<210> 4
<211> 22
<212> DNA
<213> 人工序列
<220>
<223> 针对内标的Taqman探针
<400> 4
cgcttcccgt aatccatatg ac 22
<210> 5
<211> 24
<212> DNA
<213> 人工序列
<220>
<223> 针对β-actin基因的荧光定量PCR上游引物
<400> 5
ggacctgact gactacctca tgaa 24
<210> 6
<211> 24
<212> DNA
<213> 人工序列
<220>
<223> 针对β-actin基因的Taqman探针
<400> 6
caccgagcgc ggctacagct tc 22
<210> 7
<211> 91
<212> DNA
<213> 人工序列
<220>
<223> 内标扩增产物序列
<400> 7
tgcatgcctg caggtcgact ctagaggatc tactagtcat atggattacg ggaagcggtt 60
ggtggtgcgg gaaatcgtgc gtgacattaa g 91
<210> 8
<211> 91
<212> DNA
<213> 人工序列
<220>
<223> β-actin基因扩增产物序列
<400> 8
ggacctgact gactacctca tgaagatcct caccgagcgc ggctacagct tcaccaccac 60
ggccgagcgg gaaatcgtgc gtgacattaa g 91
- 还没有人留言评论。精彩留言会获得点赞!
- 一种st2定量检测方法及试剂盒的利记博彩app
- 一种定量药粉取用盒的利记博彩app
- 一种层粘连蛋白(ln)测试试剂盒及其测试方法
- 一种抗核糖体P蛋白抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种阿胶胶液半成品或成品中驴、牛源性成分的定量检测方法、组合物及试剂盒的利记博彩app
- 一种抗PM-Scl抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种抗LKM-1抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种抗SLA/LP抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种快速定量检测h-fabp的时间分辨荧光免疫层析试剂及制备方法
- 一种定量检测液体样品中汞离子的方法及试剂盒的利记博彩app
- 一种定量药粉取用盒的利记博彩app
- 一种层粘连蛋白(ln)测试试剂盒及其测试方法
- 一种抗核糖体P蛋白抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种阿胶胶液半成品或成品中驴、牛源性成分的定量检测方法、组合物及试剂盒的利记博彩app
- 一种抗PM-Scl抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种抗LKM-1抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种抗SLA/LP抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种快速定量检测h-fabp的时间分辨荧光免疫层析试剂及制备方法
- 一种定量检测液体样品中汞离子的方法及试剂盒的利记博彩app
- 一种轴抑制蛋白2基因突变检测试剂及应用
- Ⅲ型前胶原n端肽定量测定试剂盒及其制备方法
- 一种st2定量检测方法及试剂盒的利记博彩app
- 一种定量药粉取用盒的利记博彩app
- 一种抗核糖体P蛋白抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种阿胶胶液半成品或成品中驴、牛源性成分的定量检测方法、组合物及试剂盒的利记博彩app
- 一种抗PM-Scl抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种抗LKM-1抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种抗SLA/LP抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种定量检测液体样品中汞离子的方法及试剂盒的利记博彩app
- 一种定量检测人二倍体细胞残留蛋白的试剂盒的利记博彩app
- Ⅲ型前胶原n端肽定量测定试剂盒及其制备方法
- 一种st2定量检测方法及试剂盒的利记博彩app
- 一种定量药粉取用盒的利记博彩app
- 一种抗核糖体P蛋白抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种阿胶胶液半成品或成品中驴、牛源性成分的定量检测方法、组合物及试剂盒的利记博彩app
- 一种抗PM-Scl抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种抗LKM-1抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种抗SLA/LP抗体IgG的磁微粒化学发光定量测定试剂盒及制备和检测方法
- 一种定量检测液体样品中汞离子的方法及试剂盒的利记博彩app
- 一种定量检测人二倍体细胞残留蛋白的试剂盒的利记博彩app