一种铁系催化剂及其制备方法及在异戊二烯聚合中的应用与流程
本发明涉及共轭二烯烃催化聚合技术领域,具体涉及一种铁系催化剂,还涉及所述铁系催化剂的制备方法,还涉及铁系催化剂在催化异戊二烯聚合制备高分子量及微观结构可控的聚异戊二烯中的应用。
背景技术:
异戊二烯通过1,4加成、1,2加成和3,4加成可合成顺式-1,4结构、反式-1,4结构,1,2结构及3,4结构的聚合物。微观结构差异决定了聚异戊二烯具有不同的性能。比如顺式-1,4结构聚异戊二烯的性能类似于天然橡胶,能广泛用于轮胎、胶带、胶管、胶鞋等众多橡胶加工领域,是合成橡胶中综合性能最好的一个胶种。反式-1,4结构聚异戊二烯的性能则类似于杜仲胶,此类橡胶具有优异的动态力学性能,以及良好的动态疲劳性能和耐磨性能,在所有轮胎用橡胶中滚动阻力最小。3,4聚异戊二烯则是一种高抗湿滑,低生热的合成橡胶材料,可用在轮胎胎面胶中以提高安全性能。
催化剂在聚异戊二烯结构与性能控制中起着关键作用,是聚异戊二烯合成工业的核心内容。目前工业上主要利用钛系、锂系和稀土系催化剂来合成聚异戊二烯橡胶,顺式-1,4结构一般可达98%。此外,后过渡金属催化剂由于其金属中心具备较弱的亲氧性,同时其催化得到聚合物结构呈现多样性而备受关注。其中铁系催化剂又因其价格便宜,合成简单,结构易裁剪而成为重要的研究对象。
Hiromichi最早利用铁系催化剂Fe(dmg)2/AlEt3催化异戊二烯聚合得到3,4结构为主的聚合物,但产率较低(23.2%)【J.Polym.Sci.Polym.Let.,1964,2,593-596】。Ricci报道的铁催化剂虽然产率可高达100%,但聚合物结构的选择性却不高【J.Mol.Catal.A:Chem.2003,204-205,287-293】。此外,铁系催化剂催化异戊二烯聚合,其聚合物分子量分布一般较宽且微观结构对温度特别敏感,造成合成的可控性差,得到的聚合物结构和性能不稳定,存在工业化难度【山东化工2013,42,65-66】。因此,寻求高活性并得到高分子量和微观结构可调聚合物的新型铁催化体系成为学术界的一项挑战。
长春应用化学研究所的张学全制备了芳环取代的吡啶亚胺钴配合物在AlEtCl2活化下用于1,3丁二烯聚合,获得了较高的产率和高含量的1,4结构聚丁二烯【Polyemrs 2016,8,12】。但相关的铁催化剂合成及用于异戊二烯聚合并未涉及。Ritter报道了吡啶亚胺铁配合物在烷基铝和去烷基化试剂[Ph3C]+[B(C6F5)4]-活化下催化异戊二烯高选择性聚合【Angew.Chem.Int.Ed.2012,51,11805-11808.】。取代基为辛基时得到大部分以反式-1,4结构存在的聚合物,而当取代基为芳环的三苯基苯时,得到的聚合物主要以顺式-1,4结构存在。文献中提到这种现象可能是由电子效应造成的。由于仅仅考察了两个催化剂对聚合结构的影响,很难确定催化剂构效关系,也无法对催化剂微观结构进行更大范围内的调控以使聚合物性能呈现多样性,同时去烷基化试剂[Ph3C]+[B(C6F5)4]-的使用大大提高了催化剂成本。
技术实现要素:
为了解决以上现有技术中异戊二烯聚合中使用的催化剂存在的合成的聚合物分子量分布宽、成本高、无法对催化剂微观结构进行更大范围内的调控以使聚合物性能呈现多样性的问题,本申请提供了一种新的铁系催化剂,吡啶亚胺铁配合物用作主催化剂,在已经工业化的助催化剂甲基铝氧烷(MAO)活化下,催化异戊二烯聚合,表现出较高的催化活性,并且得到了高分子量和微观结构可调的聚合物,聚合物的微观结构可通过主催化剂结构的裁剪进行调控,且对反应温度不敏感。
本发明还提供了所述铁系催化剂的制备方法。
本发明还提供了所述铁系催化剂异戊二烯聚合中的应用。
本发明是通过以下步骤得到的:
一种铁系催化剂,由主催化剂和助催化剂组成,
主催化剂为吡啶亚胺铁配合物,其结构通式如下:
式中,X为卤素(Cl或Br);R1为R2为氢(-H)、甲基(-CH3)、乙基(-CH2CH3)或苯基(-Ph);R3为氢(-H)、烃基(甲基等)、芳基(苯基、萘基等)或卤素(Br,Cl等);该配合物通过调控结构,尤其是亚胺上氮连接的取代基的电子效应和位阻效应,可增加催化剂的催化活性,同时能够有效提高聚合物的分子量并实现聚合物微观结构的调控;
助催化剂为甲基铝氧烷,结构通式为其中n为4-40的自然数;
助催化剂中的铝元素与主催化剂中的铁元素的摩尔比为500:1。
所述的铁系催化剂,优选吡啶亚胺铁配合物中X为Br或Cl,R2为-H;R3为-H。
所述的铁系催化剂,优选吡啶亚胺铁配合物结构式中X为Cl;R1为金刚烷基、三苯甲基或2,6-二(二苯甲基)-4-甲基苯基;R2为氢(-H);R3为氢(-H);结构式为
所述的铁系催化剂中主催化剂吡啶亚胺铁配合物的制备方法,在室温下,将吡啶亚胺配体在二氯甲烷溶剂中与铁的卤化物以等摩尔比反应,经过后处理,即得。后处理操作为过滤得到产物并用正己烷洗涤三次,干燥至恒重。
所述的铁系催化剂在异戊二烯聚合中的应用。
所述的应用,优选将助催化剂,甲苯和异戊二烯分别加入后,再加入主催化剂的二氯甲烷溶液进行聚合反应从而得聚异戊二烯。
所述的应用,优选单体异戊二烯与铁系催化剂中主催化剂中铁元素的摩尔比为2500:1。
所述的应用,优选聚合反应的温度为-25~25℃,聚合时间为2小时。
所述的应用,得到的聚异戊二烯数均分子量高,为6.1×104-28.2×104,分子量分布窄,为1.61-2.08。
所述的应用,根据加入主催化剂种类不同,其微观结构可调且对温度不敏感,得到的聚异戊二烯中顺式-1,4结构所占比例可调范围为62.7%~86.7%,反式-1,4结构所占比例可调范围为2.8%~8.2%,3,4结构所占比例可调范围为6.7%~34.5%。
本发明的有益效果:
1)本申请的铁系催化剂中的主催化剂制备方法简单,成本低廉;助催化剂为甲基铝氧烷(MAO),相对于背景技术中使用去烷基化试剂(Ph3C+B(C6F5)4-),更廉价易得;
2)本申请的铁系催化剂催化异戊二烯表现为较高的活性,得到的聚合物分子量高,6.1×104-28.2×104,分子量分布窄,1.61-2.08,同时聚合物的微观结构可通过主催化剂结构的裁剪进行调控,异戊二烯中顺式-1,4结构所占比例可调范围为62.7%~86.7%,反式-1,4结构所占比例可调范围为2.8%~8.2%,3,4结构所占比例可调范围为6.7%~34.5%。
3)本申请的铁系催化剂对反应温度不敏感,改变温度对聚合物微观结构影响较小,因此具有很好的工业化价值。
附图说明
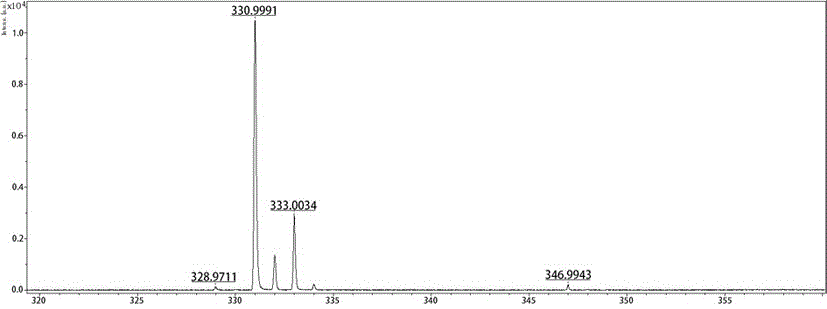
图1为实施例1得到的配合物质谱图,
图2为实施例2得到的配合物质谱图,
图3为实施例3得到的配合物质谱图。
具体实施方式
下面结合具体实施例对本发明进行进一步说明:
一、制备主催化剂吡啶亚胺铁配合物
实施例1
本实施例制备式(I)所示吡啶亚胺铁配合物:
氮气氛围下,在干燥的希莱克(Schlenk)管里加入等摩尔比的金刚烷基取代的吡啶亚胺配体和无水FeCl2,再加入10mlCH2Cl2在室温(约20℃,以下同)下搅拌24小时,产物过滤并用10ml正己烷洗涤,洗涤三次,真空干燥至恒重,得到橙色固体0.35g,即本实施例的目标产物,收率95%。
质谱分析:理论值:C16H20ClFeN2:331.0664,实测值:330.9991[M-Cl]+(图1)。
元素分析结果:理论值:C16H20Cl2FeN2:C,52.35%;H,5.49%;N,7.63%;实测值:C,52.58%;H,5.34%;N,7.38%。
红外分析:IR(KBr)/cm-1:1588,ν(C=N)。
实施例2
本实施例所制备的式(II)所示吡啶亚胺铁配合物,其中R1′=H、R2′=H,制备过程具体如下:氮气氛围下,在干燥的希莱克(Schlenk)管里加入等摩尔比的三苯甲基取代的吡啶亚胺配体和无水FeCl2,再加入10mlCH2Cl2在室温下搅拌24小时,产物过滤并用10ml正己烷洗涤,洗涤三次,真空干燥至恒重,得到浅粉色固体0.46g,即本实施例的目标产物,收率96%。质谱分析:理论值:C25H20ClFeN2:439.0664,实测值:439.0714[M-Cl]+(图2)。
元素分析结果:理论值:C25H20Cl2FeN2:C,63.19%;H,4.24%;N,5.90%;实测值:C,62.92%;H,4.19%;N,5.72%。
红外分析:IR(KBr)/cm-1:1587,ν(C=N)。
实施例3
本实施例所制备的式(III)所示吡啶亚胺铁配合物,其中R1"=H、R2"=H、R3"=CH3,制备过程具体如下:
氮气氛围下,在干燥的希莱克(Schlenk)管里加入等摩尔比的2,6-二(二苯甲基)-4-甲基苯基取代的吡啶亚胺配体和无水FeCl2,再加入10mlCH2Cl2在室温下搅拌24小时,产物过滤并用10ml正己烷洗涤,洗涤三次,真空干燥至恒重,得到酒红色固体0.61g,即本实施例的目标产物,收率93%。
质谱分析:理论值:C39H32ClFeN2:619.1603,实测值:619.0020[M-Cl]+(图3)。
元素分析结果:理论值:C39H32Cl2FeN2:C,71.47%;H,4.92%;N,4.27%;实测值:C,71.98%;
H,4.88%;N,4.18%。
红外分析:IR(KBr)/cm-1:1593,ν(C=N)。
二、异戊二烯聚合反应
实施例4
在氮气氛围下,在50ml希莱克(Schlenk)管中,加入三异丁基铝(TIBA)4mmol,无水甲苯7ml,异戊二烯2ml(20.0mmol),用1ml CH2Cl2溶解的实施例1制备得到的催化剂2.9mg(8μmol),在25℃下聚合2h,反应用20ml的乙醇稀盐酸(50:1)溶液终止,未得到聚合物。在使用助催化剂三异丁基铝情况下无聚合活性。
实施例5
在氮气氛围下,在50ml希莱克(Schlenk)管中,加入氯化二乙基铝(AlEt2Cl)4mmol,无水甲苯7ml,异戊二烯2ml(20.0mmol),用1ml CH2Cl2溶解的实施例1制备得到的催化剂2.9mg(8μmol),在25℃下聚合2h,反应用20ml的乙醇稀盐酸(50:1)溶液终止,未得到聚合物。在使用助催化剂氯化二乙基铝情况下无聚合活性。
实施例6
在氮气氛围下,在50ml希莱克(Schlenk)管中,加入二氯化乙基铝(AlEtCl2)4mmol,无水甲苯7ml,异戊二烯2ml(20.0mmol),用1ml CH2Cl2溶解的实施例1制备得到的催化剂2.9mg(8μmol),在25℃下聚合2h,反应用20ml的乙醇稀盐酸(50:1)溶液终止,过滤,用乙醇洗三次,室温下真空干燥至恒重,产率:53.4%,数均分子量:1800,多分散系数:4.70。在使用助催化剂二氯化乙基铝情况下所得聚合物分子量为1800,远低于实施例9中用甲基铝氧化作助催化剂时所得聚合物的分子量(7.0×104)。
实施例7(实施例7-9是筛选主催化剂与助催化剂的比例,分别为200:1;800:1;500:1)
在氮气氛围下,在50ml希莱克(Schlenk)管中,加入0.09g(1.6mmol)甲基铝氧烷(MAO),无水甲苯7ml,异戊二烯2ml(20.0mmol),用1ml CH2Cl2溶解的实施例1制备得到的催化剂2.9mg(8μmol),在25℃下聚合2h,反应用20ml的乙醇稀盐酸(50:1)溶液终止,过滤,用乙醇洗三次,室温下真空干燥至恒重,产率:12.1%,数均分子量:3.1×104,多分散系数():2.63。产率较低不作为优选对象。
实施例8
在氮气氛围下,在50ml希莱克(Schlenk)管中,加入0.36g(6.4mmol)甲基铝氧烷(MAO),无水甲苯7ml,异戊二烯2ml(20.0mmol),用1ml CH2Cl2溶解的实施例1制备得到的催化剂2.9mg(8μmol),在25℃下聚合2h,反应用20ml的乙醇稀盐酸(50:1)溶液终止,过滤,用乙醇洗三次,室温下真空干燥至恒重,产率:40.1%,数均分子量:1.3×104,多分散系数:2.07。产率亦不高,不作为优选对象。
实施例9
在氮气氛围下,在50ml希莱克(Schlenk)管中,加入0.23g(4mmol)甲基铝氧烷(MAO),无水甲苯7ml,异戊二烯2ml(20.0mmol),用1ml CH2Cl2溶解的实施例1制备得到的催化剂2.9mg(8μmol),在25℃下聚合2h,反应用20ml的乙醇稀盐酸(50:1)溶液终止,过滤,用乙醇洗三次,室温下真空干燥至恒重,产率:58.2%,数均分子量:7.0×104,多分散系数:1.82。不同结构所占比例:顺式-1,4结构占76.8%,反式-1,4结构占8.2%(顺式-1,4结构与反式-1,4结构之比为90:10),3,4结构占15.0%。
实施例10
聚合反应所用实施例1制备得到的催化剂2.9mg(8μmol),其它同实施例9,反应温度为-25℃,产率:46.2%,数均分子量:11.2×104,多分散系数:1.78。不同结构所占比例:顺式-1,4结构占83.4%,反式-1,4结构占6.2%(顺式-1,4结构与反式-1,4结构之比为91:9),3,4结构占10.4%。
实施例11
聚合反应所用实施例1制备得到的催化剂2.9mg(8μmol),其它同实施例9,反应温度为0℃,产率:51.2%,数均分子量:8.9×104,多分散系数:1.81。不同结构所占比例:顺式-1,4结构占78.4%,反式-1,4结构占7.3%(顺式-1,4结构与反式-1,4结构之比为91:8),3,4结构占14.3%。
根据实施例9-11可发现温度对聚合物微观结构的影响较小,在-25-25℃下,顺式-1,4结构比例变化不大,为76.8~83.4%。
实施例12
聚合反应所用实施例2制备得到的催化剂3.8mg(8μmol),其它同实施例9,反应温度为-25℃,产率:50.1%,数均分子量:12.3×104,多分散系数:2.03。不同结构所占比例:顺式-1,4结构占86.7%,反式-1,4结构占6.6%(顺式-1,4结构与反式-1,4结构之比为91:9),3,4结构占6.7%。
实施例13
聚合反应所用实施例2制备得到的催化剂3.8mg(8μmol),其它同实施例9,反应温度为0℃,产率:53.6%,数均分子量:9.6×104,多分散系数:2.07。不同结构所占比例:顺式-1,4结构占83.2%,反式-1,4结构占7.0%(顺式-1,4结构与反式-1,4结构之比为91:10),3,4结构占9.8%。
实施例14
聚合反应所用实施例2制备得到的催化剂3.8mg(8μmol),其它同实施例9,反应温度为25℃,产率:61.3%,数均分子量:6.1×104,多分散系数:2.08。不同结构所占比例:顺式-1,4结构占78.2%,反式-1,4结构占7.6%(顺式-1,4结构与反式-1,4结构之比为91:9),3,4结构占14.2%。
实施例15
聚合反应所用实施例3制备得到的催化剂5.2mg(8μmol),其它同实施例9,反应温度为-25℃,产率:73.2%,数均分子量:28.2×104,多分散系数:1.70。不同结构所占比例:顺式-1,4结构占69.9%,反式-1,4结构占4.5%(顺式-1,4结构与反式-1,4结构之比为94:6),3,4结构占25.6%。
实施例16
聚合反应所用实施例3制备得到的催化剂5.2mg(8μmol),其它同实施例9,反应温度为0℃,产率:79.7%,数均分子量:21.6×104,多分散系数:1.63。不同结构所占比例:顺式-1,4占63.9%,反式-1,4结构占3.0%(顺式-1,4结构与反式-1,4结构之比为96:4),3,4结构占33.1%。
实施例17
聚合反应所用实施例3制备得到的催化剂5.2mg(8μmol),其它同实施例9,反应温度为25℃,产率:85.7%,数均分子量:18.2×104,多分散系数:1.61。不同结构所占比例:顺式-1,4结构占62.7%,反式-1,4结构占2.8%(顺式-1,4结构与反式-1,4结构之比为96:4),3,4结构占34.5%。
各实施例的数据设置如下表。
由实施例9,14,17可知,调控R1取代基可使得聚合物順式-1,4结构比例从62.7%增加到78.2%,3,4结构比例从34.5减少到8.2%。
通过上述比较实例可以看出,本发明的主催化剂随着其亚胺上氮连接的取代基的电子效应及位阻效应不同,可调控所得聚合物的分子量及微观结构,吸电子取代基和大位阻取代基有利于分子量的提高和3,4结构的增加。此外,改变温度对聚合物微观结构影响较小。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受实施例的限制,其它任何未背离本发明的精神实质与原理下所做的改变、修饰、组合、替代、简化均应为等效替换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!