2‑(3‑溴‑4‑(3‑氟苄氧基)苯基)‑1,3‑二氧戊环及其制备方法与流程
本发明涉及一种新化合物2-(3-溴-4-(3-氟苄氧基)苯基)-1,3-二氧戊环,本发明还涉及该化合物的制备方法。
背景技术:
沙芬酰胺(Safinamide),化学名为(S)-2-[4-(3-氟苄氧基)苄氨基]丙酰胺(式I),是纽朗制药公司研发的用于一种用于治疗帕金森病的新型药物。
目前,合成沙芬酰胺的方法:对羟基苯甲醛与间氟氯苄在一定反应条件下反应,得到中间产物4-(3-氟苄氧基)苯甲醛(中间体-1);然后,L-丙氨酰胺盐酸盐和中间体-1反应生成(S)-2-[4-(3-氟苄氧基)苄氨基]丙酰胺(中间体-2);最后,还原中间体-2得到沙芬酰胺。
已公开的制备沙芬酰胺的工艺中,3-(3-氟苄基)-4-(3-氟苄氧基)苯甲醛(式9)结构式如下。该杂质是一种潜在的关键中间体杂质,更早的研究、控制杂质,一方面提高了沙芬酰胺的质量标准,另一方面确保制备出符合质量标准的产品,对于沙芬酰胺的研究具有重要的意义。
已公开的制备工艺中,对羟基苯甲醛与过量的间氟氯苄在碳酸钾、甲苯存在下,高温反应时间可长达5天,反应物料的气相色谱分析显示,此时的反应物料由4-(3-氟苄氧基)苯甲醛(中间体-1)和3-(3-氟苄基)-4-(3-氟苄氧基)苯甲醛(式9)以91.4:8.6(面积比)比例混合;从该混合物料中通过分馏获得杂质9,收率低至3.6%。
目前现有技术中并没有直接制备杂质9的方法,只是在制备沙芬酰胺的过程中将其分离出来,收率极低。
为了解决上述问题,本发明提供了一种新化合物2-(3-溴-4-(3-氟苄氧基)苯基)-1,3-二氧戊环,该化合物可用于制备3-(3-氟苄基)-4-(3-氟苄氧基)苯甲醛,并获得理想的收率。
技术实现要素:
本发明所要解决的技术问题是针对现有技术的现状,提供一种新化合物2-(3-溴-4-(3-氟苄氧基)苯基)-1,3-二氧戊环,该化合物可用于制备3-(3-氟苄基)-4-(3-氟苄氧基)苯甲醛,并使收率显著提高。
本发明所要解决的另一个技术问题是针对现有技术的现状,提供一种2-(3-溴-4-(3-氟苄氧基)苯基)-1,3-二氧戊环的制备方法。
本发明解决上述技术问题所采用的技术方案为:一种新化合物2-(3-溴-4-(3-氟苄氧基)苯基)-1,3-二氧戊环,其特征在于结构式如下:
一种上述新化合物2-(3-溴-4-(3-氟苄氧基)苯基)-1,3-二氧戊环的制备方法,其特征在于包括以下步骤:
(1)制备3-溴-4-(3-氟苄氧基)苯甲醛
在碳酸钾、四丁基溴化铵存在下,向有机溶剂中加入3-溴-4-羟基苯甲醛(下式1化合物)及间氟溴苄(下式2化合物),反应后,得到3-溴-4-(3-氟苄氧基)苯甲醛(下式3化合物);
(2)制备4-甲基苯磺酸吡啶鎓
对甲苯磺酸(下式4化合物)与吡啶(下式5化合物)在氮气保护下,反应生成4-甲基苯磺酸吡啶鎓(下式6化合物);
(3)合成2-(3-溴-4-(3-氟苄氧基)苯基)-1,3-二氧戊环
步骤(1)所得3-溴-4-(3-氟苄氧基)苯甲醛(式3化合物)与步骤(2)所得4-甲基苯磺酸吡啶鎓(式6化合物)反应,得到2-(3-溴-4-(3-氟苄氧基)苯基)-1,3-二氧戊环(下式7化合物)。
作为优选,步骤(1)在氮气保护下进行,反应温度为70~90℃。
进一步优选,反应完毕后,将反应物冷却至室温,过滤,除去无机盐,有机溶剂洗涤残留物,减压浓缩滤液,得到3-溴-4-(3-氟苄氧基)苯甲醛,提纯备用。
优选地,步骤(2)在氮气保护下进行,反应温度为0~5℃,反应完毕后,减压干燥,得到4-甲基苯磺酸吡啶鎓。
优选地,步骤(3)在乙二醇溶剂存在下,反应温度为90℃以上;反应完毕后冷却、洗涤、减压干燥得到2-(3-溴-4-(3-氟苄氧基)苯基)-1,3-二氧戊环。
与现有技术相比,本发明的优点在于:本发明提供了一种新化合物2-(3-溴-4-(3-氟苄氧基)苯基)-1,3-二氧戊环,该新化合物可用于直接制备3-(3-氟苄基)-4-(3-氟苄氧基)苯甲醛,且制备的3-(3-氟苄基)-4-(3-氟苄氧基)苯甲醛收率高达64.85%,高效液相色谱测得的纯度达到98.92%。
附图说明
图1为本发明实施例中步骤(1)产物的HPLC图谱;
图2为本发明实施例中步骤(1)产物的GC图谱;
图3为本发明实施例中步骤(1)产物的TIC图谱;
图4为本发明实施例中步骤(1)产物的UV图谱;
图5为本发明实施例中步骤(1)产物的Mass图谱;
图6为本发明实施例中步骤(1)产物的IR图谱;
图7为本发明实施例中步骤(2)产物的HPLC图谱;
图8为本发明实施例中步骤(2)产物的TIC图谱;
图9为本发明实施例中步骤(2)产物的UV图谱;
图10为本发明实施例中步骤(2)产物的Mass图谱(吡啶);
图11为本发明实施例中步骤(2)产物的Mass图谱(对-甲苯磺酸);
图12为本发明实施例中步骤(3)产物的GC图谱;
图13为本发明实施例中步骤(3)产物的TIC图谱;
图14为本发明实施例中步骤(3)产物的UV图谱;
图15为本发明实施例中步骤(3)产物的Mass图谱;
图16为本发明实施例中步骤(3)产物的IR图谱;
图17为本发明实施例中步骤(a)产物的HPLC图谱;
图18为本发明实施例中步骤(a)产物的TIC图谱;
图19为本发明实施例中步骤(a)产物的UV图谱;
图20为本发明实施例中步骤(a)产物的Mass图谱;
图21为发明实施例中步骤(a)产物的IR图谱;
图22为本发明实施例中步骤(b)产物的HPLC图谱;
图23为本发明实施例中步骤(b)产物的TIC图谱;
图24为本发明实施例中步骤(b)产物的UV图谱;
图25为本发明实施例中步骤(b)产物的Mass图谱;
图26为发明实施例中步骤(b)产物的IR图谱。
具体实施方式
以下结合附图实施例对本发明作进一步详细描述。
本实施例的新化合物2-(3-溴-4-(3-氟苄氧基)苯基)-1,3-二氧戊环结构式如下:
上述化合物的特性如下:
IR(KBr;cm-1):2886(C-H),1591(C=C),1086(C-O-C-O),1043(Ar-O-C),552(C-Br);
1H NMR(400MHz,DMSO-d6,δppm):7.61-7.62(d,1H,Ar-H),7.39-7.45(m,1H,Ar-H),7.36-7.39(dd,1H,Ar-H),7.26-7.39(m,2H,Ar-H),7.17-7.24(d,1H,Ar-H),7.11-7.16(m,1H,Ar-H),5.65(s,1H,O-CH-O),5.22(s,2H,O-CH2),3.99-4.02(m,2H,O-CH2-CH2-O),3.87-3.90(m,2H,O-CH2-CH2-O);
13C NMR(400MHz,DMSO-d6,δppm):163.83,155.13,139.85,132.74,131.56,131.00,127.86,123.55,115.10,114.40,114.14,111.36,102.19,69.65,65.25;
MS(ESI+ve,m/z,Exact mass=352.01):352.9(M+H),307,289,273,124。
本实施例的新化合物2-(3-溴-4-(3-氟苄氧基)苯基)-1,3-二氧戊环的制备方法包括以下步骤:
步骤一:依次向反应器中加入3-溴-4-羟基苯甲醛(25g,0.12mol)、甲苯(250mL)、碳酸钾(22.2g,0.161mol)、四丁基溴化铵(20g,0.062mol),然后通入氮气,所得混合物在室温下搅拌;
在氮气保护下,向上述反应混合物中滴加间氟溴苄(23.5g,0.124mol),滴加完毕后,将该混合物缓慢加热到70~90℃,保温反应6小时;反应过程中由薄层色谱分析法检测(流动相为正己烷和10%的乙酸乙酯,在254nm紫外灯下),如果薄层色谱反应不明显,再加入间氟溴苄(4.5g,0.023mol),直至反应进行到薄层色谱显色良好;
反应完毕后,将反应物冷却至室温,过滤除去无机盐,残留固相用25mL甲苯洗涤,在50~55℃的减压条件下浓缩滤液,得到3-溴-4-(3-氟苄氧基)苯甲醛粗品;
提纯:3-溴-4-(3-氟苄氧基)苯甲醛粗品在25~30℃下加入甲苯(25mL)搅拌溶解,在搅拌下,向溶液中滴加正己烷(50mL),得到的悬浮液冷却至0~5℃并放置1小时;过滤固体,滤饼用正己烷洗涤(25mL),将湿的滤饼放在45~50℃的减压烘箱中干燥4小时,得到3-溴-4-(3-氟苄氧基)苯甲醛。
如图1~6所示,所得3-溴-4-(3-氟苄氧基)苯甲醛的质量为31g,收率为80.6%,GC色谱测得纯度为99.66%,HPLC测得纯度为99.34%。
步骤二:向反应器中加入对甲苯磺酸(5g,0.029mol),在氮气保护下,继续加入四氢呋喃(40mL),室温反应;将反应溶液冷却至0~5℃,然后,向溶液中滴加吡啶(2.75g,0.035mol),并保证20~30分钟内滴加完,然后搅拌30分钟;
在氮气保护下过滤,所得滤饼用5mL四氢呋喃洗涤,将其放在45~50℃的减压烘箱中干燥4小时,得到4-甲基苯磺酸吡啶鎓。
如图7~11所示,所得4-甲基苯磺酸吡啶鎓质量为6g,收率82.19%,HPLC检测纯度100%。
步骤三:向反应器中加入3-溴-4-(3-氟苄氧基)苯甲醛(25g,0.081mol),在搅拌下,加入甲苯(500mL)、乙二醇(10g,0.161mol)、4-甲基苯磺酸吡啶鎓(1g,0.04mol);将反应混合物缓慢加热至回流,蒸出溶剂,反应溶液在回流温度下与水共沸蒸馏5小时,气相色谱检测反应,如果反应未按计划进行,再加入乙二醇(5g,0.081mol)保证反应继续;反应结束后,冷却、50mL NaHCO3溶液洗涤;洗涤后的产物在5.0g硫酸钠中干燥,在25~30℃温度下,减压浓缩得到2-(3-溴-4-(3-氟苄氧基)苯基)-1,3-二氧戊环。
如图12~16所示,所得化合物7质量为27.5g,收率96.3%,气相色谱检测纯度为98.89%。
采用本实施例的新化合物2-(3-溴-4-(3-氟苄氧基)苯基)-1,3-二氧戊环制备3-(3-氟苄基)-4-(3-氟苄氧基)苯甲醛的步骤如下:
(a)向反应器中加入化合物7(25g,0.071mol),随后加入四氢呋喃(375mL)和硼酸三异丙酯(24g,0.127mol),在氮气保护下常温反应;混合物冷却至-78℃,在该温度下缓慢滴加正丁基锂-正己烷混合物(11.3g,0.176mol),在氮气保护下反应1小时,滴加完毕后搅拌反应4小时;薄层层析色谱(TLC)检测反应进度,如果检测到反应不理想,再加入正丁基锂-正己烷混合物(1g,0.015mol),维持反应继续;
反应结束后,用稀盐酸(1:1)调节pH至1~2,反应混合物加热至25~30℃,再加入25ml水,搅拌30分钟;乙酸乙酯萃取(3×75mL),硫酸钠干燥有机相(10g),在50~55℃下减压浓缩,获得粗品2-(3-氟苄氧基)-5-甲酰基苯硼酸。
提纯:在室温下,2-(3-氟苄氧基)-5-甲酰基苯硼酸粗品中加入二氯甲烷(50mL),搅拌下滴加正己烷(250mL),得到的悬浮液冷却至0~5℃并放置1小时;过滤,正己烷洗涤滤饼(25mL),将湿的滤饼放置到减压烘箱中干燥4小时,得到2-(3-氟苄氧基)-5-甲酰基苯硼酸。
如图17~21所示,所得2-(3-氟苄氧基)-5-甲酰基苯硼酸质量为13.8g,收率71.13%,HPLC检测纯度96.19%。
(b)向反应器中加入2-(3-氟苄氧基)-5-甲酰基苯硼酸(25g,0.91mol),在室温、氮气条件下,加入1,4-二恶烷(250mL)、水(25mL)、间氟溴苄(25.8g,0.136mol),然后,通30分钟氮气;再加入Pd2(dba)3(1.75g,0.002mol),缓慢升温至70℃,搅拌反应两小时,通过薄层色谱检测反应;
反应结束后,混合物冷却至室温,用硅藻土过滤,1,4-二恶烷(25mL)洗涤滤饼;然后在旋转蒸发仪60~65℃下减压蒸馏得到油相;残余的油相加入水(100ml),再用乙酸乙酯萃取(3×100mL),硫酸钠(10g)干燥有机相,在50~55℃减压干燥粗品,得到3-(3-氟苄基)-4-(3-氟苄氧基)苯甲醛;
提纯:3-(3-氟苄基)-4-(3-氟苄氧基)苯甲醛粗品经硅胶柱层析法(mesh-20)(洗脱液:正己烷:乙酸乙酯(9.8:0.2))纯化。
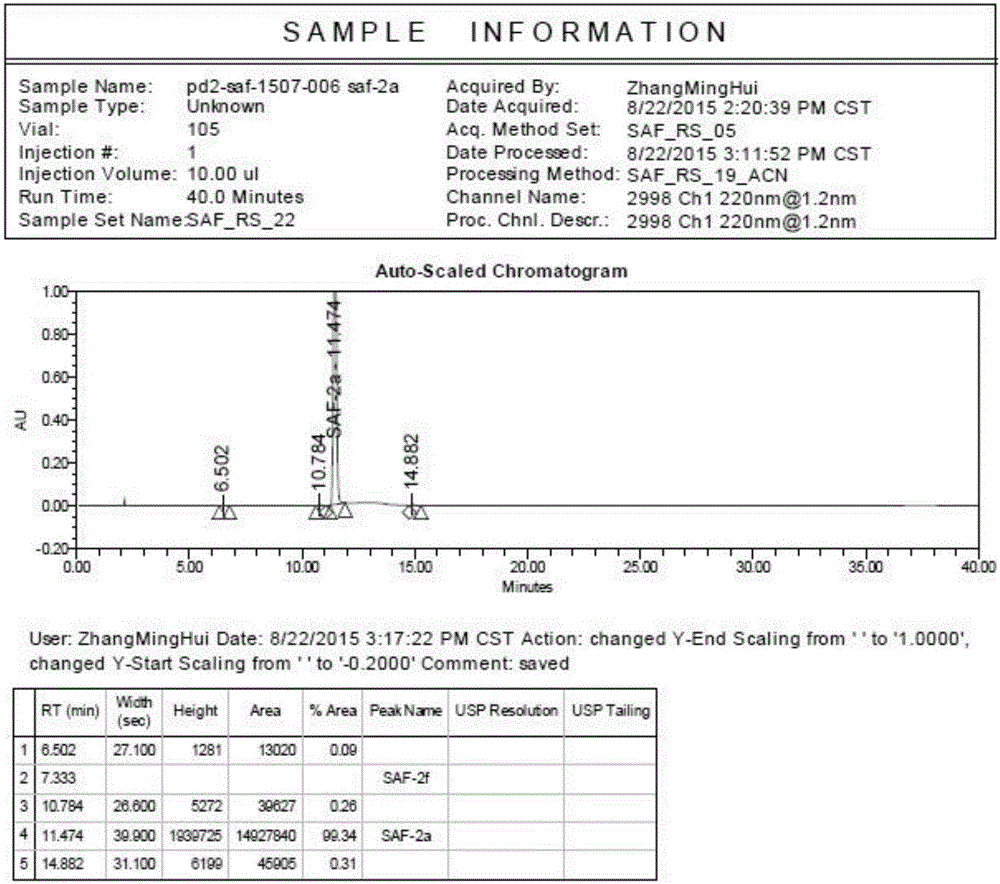
如图22~26所示,所得3-(3-氟苄基)-4-(3-氟苄氧基)苯甲醛质量为20g,收率为64.85%,HPLC检测纯度为97.82%。
可见,与现有技术中制备3-(3-氟苄基)-4-(3-氟苄氧基)苯甲醛的方法相比,本实施例采用新化合物2-(3-溴-4-(3-氟苄氧基)苯基)-1,3-二氧戊环制备3-(3-氟苄基)-4-(3-氟苄氧基)苯甲醛的收率得到显著提高。
- 还没有人留言评论。精彩留言会获得点赞!