牛樟芝素的制备方法与流程
本发明涉及一种牛樟芝素的制备方法。
背景技术:
antrocin(牛樟芝素)是由姜宏哲(hung-chenchiang)教授和他的合作者于1994年从中国台湾牛樟芝(又名牛樟菇,非褶菌目、多孔科、多年生蕈菌类,学名为antrodiacamphorata)中分离出来的,分子结构式为
牛樟芝的活性成分三萜类化合物除个别特殊外在其他薄孔菌属的真菌中也均有发现。但是倍半萜化合物牛樟芝素却唯有在野生牛樟芝的子实体中才能分离到,人工手段培养的牛樟芝尚未有报道分离得到牛樟芝素。
从结构上看,牛樟芝素是一种倍半萜稀内酯,具备一个6.6.5三环结构,包含三个手性中心及两个全碳取代中心。2011年,台湾朝阳科技大学的曾耀铭教授对牛樟芝素的生物活性进行研究。研究发现该分子具有选择性抑制人转移性乳腺癌细胞mda-mb-231细胞系的生理活性,其ic50值为0.6μm。2013年,曾耀铭教授又发现牛樟芝素在抑制人非小细胞肺癌细胞有很好的抑制增殖作用,对具野生型表皮生长因子受体(egfr)的肺癌细胞h441的ic50值为0.75μm,而对具突变型egfr的h1975肺癌细胞的ic50值0.83μm。
牛樟芝素具有良好的潜在抗癌活性,目前的人工合成报道,而且还未见有不对此合成的报道,因此目前的合成技术尚无法提供充足的牛樟芝素,以对其进行深入的活性及构效关系、毒理病理研究。因此,急需找到一种可以大量合成牛樟芝素的方法,以便提供足够的牛樟芝素来研究其病理毒理、构效关系,并且通过结构修饰来阐明和改进它的生物活性。
技术实现要素:
基于此,有必要提供一种可以大量合成的牛樟芝素的牛樟芝素的制备方法,通过以化合合成手段大量的得到牛樟芝素,以便对牛樟芝素进行结构修饰和活性研究。
一种牛樟芝素的制备方法,包括如下步骤:
步骤一、将结构式为
步骤二、向所述第一反应液中加入硼氢化钠,剧烈搅拌并充分反应,反应完成后纯化得到结构式为
步骤三、在-78℃的条件下,将所述化合物a溶解于第二有机溶剂中,分批次投入碎钠块,搅拌并充分反应,得到含有结构式为
步骤四、除去所述第二反应液中的有机溶剂后酸化并萃取所得产物,接着除去萃取剂并加入第三有机溶剂,然后加入对甲苯磺酸,于60℃~100℃下充分反应,反应完成后纯化得到结构式为
步骤五、将所述化合物d溶解于第四有机溶剂中,加入碘、三苯基膦和咪唑,搅拌并充分反应,反应完成后纯化得到结构式为
步骤六、将所述化合物d1溶解于第五有机溶剂中,加入第一有机碱,在80℃下搅拌并充分反应,反应完成后纯化得到牛樟芝素。
在一个实施例中,步骤一中,所述第一有机溶剂为二氯甲烷、甲醇或体积比为1~6:1的二氯甲烷和甲醇的混合液体;
步骤一中,所述鼓气通入臭氧的操作的时间为1h。
在一个实施例中,所述第一有机溶剂为体积比为3:1的二氯甲烷和甲醇的混合液体、体积比为4:1的二氯甲烷和甲醇的混合液体或体积比为5:1的二氯甲烷和甲醇的混合液体。
在一个实施例中,步骤二中,所述剧烈搅拌并充分反应的操作的时间为1h。
在一个实施例中,步骤三中还包括在所述分批次投入碎钠块的操作之后,加入低元醇的操作,所述低元醇的碳原子数为1~4。
在一个实施例中,步骤三中,所述第二有机溶剂为液氨或体积比为5~10:1的液氨与四氢呋喃的混合液体;
步骤三中,所述剧烈搅拌并充分反应的操作的时间为1h。
在一个实施例中,步骤四中,所述酸化的操作所用的酸为盐酸或醋酸,所述酸化的操作为酸化至ph为1~2;
步骤四中,所述萃取的操作中,所述萃取剂为乙酸乙酯或二氯甲烷。
在一个实施例中,步骤四中,所述第三有机溶剂为甲苯、苯或四氢呋喃;
步骤四中,所述100℃下充分反应的时间为1h~3h。
在一个实施例中,步骤五中,所述第四有机溶剂为四氢呋喃或二氯甲烷;
步骤五中,所述搅拌并充分反应的操作的时间为1h~3h。
在一个实施例中,步骤六中,所述第一有机碱为1,8-二氮杂二环十一碳-7-烯或乙醇钠;步骤六中,所述在80℃下搅拌并充分反应的操作的时间为3h。
这种牛樟芝素的制备方法操作简单,步骤简短,易于工业化生产,通过步骤一的臭氧氧化切断反应、步骤三的伯奇还原反应、步骤四酯缩合反应以及步骤五和通过步骤六的碘代-消除反应,快速地构建了分子的骨架结构,从而完成了牛樟芝素的不对称全合成。这种牛樟芝素的制备方法可以大量合成的牛樟芝素,合成的牛樟芝素可以用来研究其病理毒理、构效关系,并且通过结构修饰来阐明和改进它的生物活性。
附图说明
图1为一实施方式的牛樟芝素的制备方法的制备方法的流程图。
具体实施方式
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合附图和具体实施例对本发明的具体实施方式做详细的说明。在下面的描述中阐述了很多具体细节以便于充分理解本发明。但是本发明能够以很多不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似改进,因此本发明不受下面公开的具体实施的限制。
本发明的牛樟芝素的制备方法,其各步反应均为在氮气保护下、干燥的溶剂中、无水的条件下进行。
本发明的牛樟芝素的制备方法中,所有使用的化学试剂都是商业可得的原料,无须进一步处理。
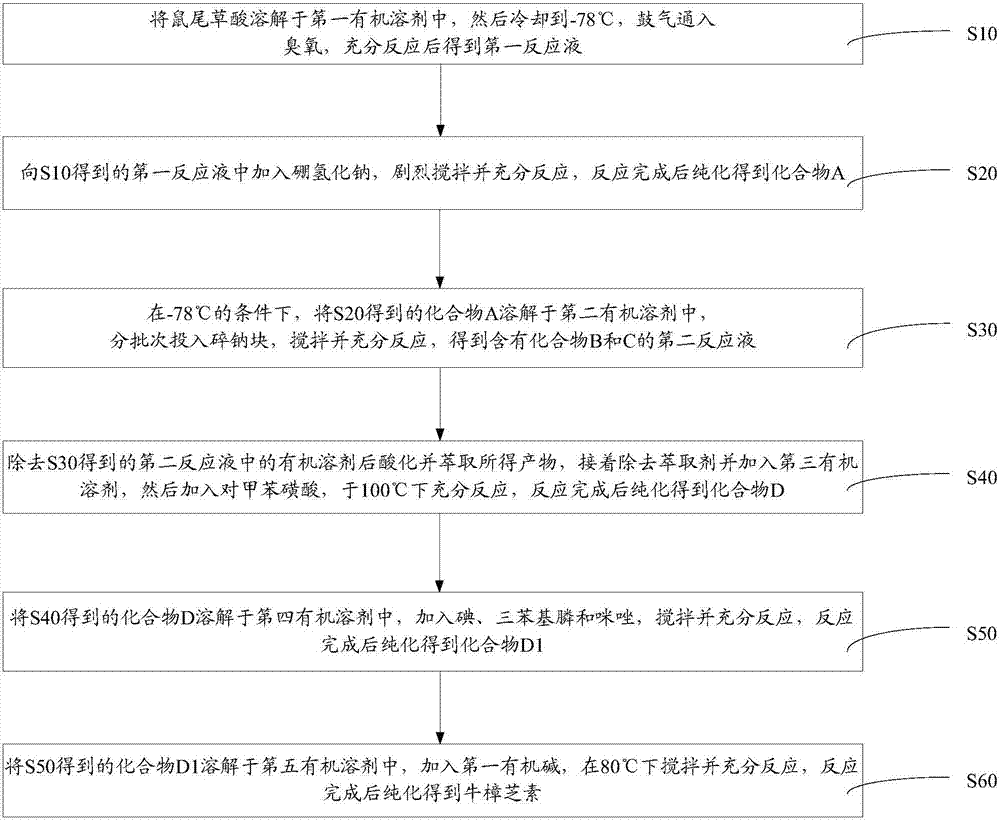
如图1所示的一实施方式的牛樟芝素的制备方法,包括如下步骤:
s10、将鼠尾草酸溶解于第一有机溶剂中,然后冷却到-78℃,鼓气通入臭氧,充分反应后得到第一反应液。
鼠尾草酸的结构式为
s10中,第一有机溶剂可以为二氯甲烷、甲醇或体积比为2~6:1的二氯甲烷和甲醇的混合液体。
s10中,鼓气通入臭氧的操作的时间为1h。
优选的,第一有机溶剂为体积比为3:1的二氯甲烷和甲醇的混合液体、体积比为4:1的二氯甲烷和甲醇的混合液体或体积比为5:1的二氯甲烷和甲醇的混合液体。
特别优选的,第一有机溶剂为体积比为3:1的二氯甲烷和甲醇的混合液体。
s20、向s10得到的第一反应液中加入硼氢化钠,剧烈搅拌并充分反应,反应完成后纯化得到化合物a。
化合物a的结构式为
s20中,鼠尾草酸与硼氢化钠的摩尔比为1:3~10。
s20中,剧烈搅拌并充分反应的操作的时间为1h。
s20中,反应完成后纯化得到化合物a的操作为:加入饱和的氯化铵溶液淬灭反应,用乙酸乙酯萃取,有机相用饱和氯化钠溶液洗涤、无水硫酸钠干燥,过滤后真空浓缩,然后用硅胶柱层析纯化,得到化合物a。
s30、在-78℃的条件下,将s20得到的化合物a溶解于第二有机溶剂中,分批次投入碎钠块,搅拌并充分反应,得到含有化合物b和c的第二反应液。
化合物b结构式为
s30中,化合物a与碎钠块的摩尔比为1:5~20;
s30中,第二有机溶剂为液氨或体积比为5:1~10:1的液氨与四氢呋喃的混合液体。
优选的,第二有机溶剂为液氨。
s30中,剧烈搅拌并充分反应的操作的时间为1h。
s30中还包括在分批次投入碎钠块的操作之后,加入低元醇的操作。低元醇的碳原子数为1~4。
低元醇与化合物a的摩尔比为5~10:1。
低元醇可以为甲醇、乙醇、叔丁醇或异丙醇。
优选的,低元醇可以为乙醇。
s40、除去s30得到的第二反应液中的有机溶剂后酸化并萃取所得产物,接着除去萃取剂并加入第三有机溶剂,然后加入对甲苯磺酸,于100℃下充分反应,反应完成后纯化得到化合物d。
化合物d的结构式为
s40中,对甲苯磺酸和化合物a的摩尔比为0.1~0.5:1。
s40中,酸化的操作所用的酸为盐酸或醋酸,酸化的操作为酸化至ph为1~2。
优选的,酸化的操作所用的酸为盐酸。
s40中,萃取的操作中,萃取剂为乙酸乙酯或二氯甲烷。
优选的,萃取剂为乙酸乙酯。
s40中,第三有机溶剂为甲苯、苯或四氢呋喃;
s40中,100℃下充分反应的时间为1h~3h。
s40中的反应,在得到化合物d的同时还会得到结构式为
通过在s30中加入低元醇,可以增加s40得到的反应物中化合物d的比例,提高了产率。
s40中,反应完成后纯化得到化合物d的操作为:加入饱和碳酸氢钠溶液,用乙酸乙酯萃取,有机相用饱和氯化钠溶液洗涤、无水硫酸钠干燥,过滤后真空浓缩,然后用硅胶柱层析纯化,得到化合物d。
s50、将s40得到的化合物d溶解于第四有机溶剂中,加入碘、三苯基膦和咪唑,搅拌并充分反应,反应完成后纯化得到化合物d1。
化合物d1的结构式为
s50中,碘与化合物d的摩尔比为1.1~1.6:1,三苯基膦与化合物d的摩尔比为1~1.3:1,咪唑与化合物d的摩尔比为1.2~1.7:1。
s50中,第四有机溶剂为四氢呋喃或二氯甲烷。
优选的,第四有机溶剂为四氢呋喃。
s50中,搅拌并充分反应的操作的时间为3h。
反应完成后纯化得到化合物d1的操作为:加入饱和氯化铵溶液淬灭反应,用乙酸乙酯萃取,有机相用饱和氯化钠溶液洗涤、无水硫酸钠干燥,过滤后真空浓缩,然后用硅胶柱层析纯化,得到化合物d1。
s60、将s50得到的化合物d1溶解于第五有机溶剂中,加入第一有机碱,在80℃下搅拌并充分反应,反应完成后纯化得到牛樟芝素。
s60中,第五有机溶剂为甲苯、苯或乙醇。
s60中,第一有机碱为1,8-二氮杂二环十一碳-7-烯或乙醇钠。
优选的,第一有机碱为1,8-二氮杂二环十一碳-7-烯。
s60中,在80℃下搅拌并充分反应的操作的时间为3h。
这种牛樟芝素的制备方法操作简单,步骤简短,易于工业化生产,通过s10的臭氧氧化切断反应、s30的伯奇还原反应、s40酯缩合反应以及s50和s60的碘代-消除反应,快速地构建了分子的骨架结构,从而完成了牛樟芝素的不对称全合成。这种牛樟芝素的制备方法可以大量合成的牛樟芝素,合成的牛樟芝素可以用来研究其病理毒理、构效关系,并且通过结构修饰来阐明和改进它的生物活性。
以下为具体实施例。
实施例中,反应检测时使用的是0.25mm烟台银龙硅胶有限公司生产的薄层色谱硅胶板(60f-254);柱层析使用青岛谱科分离材料有限公司生产的200-300目硅胶,使用石油醚沸程为60~90℃;核磁共振数据由以下仪器测得:brükeradvance500或brükeradvance400或brükeradvance300,使用tms或氘代溶剂中残留的未氘代溶剂做内标,解释多重裂分时所用到的简写为s单峰,d双重裂分,t三重裂分,q四重裂分,m多重裂分,br宽峰;质谱由absciex
实施例1
在无水的条件下,将结构式为
在第一反应液中加入硼氢化钠(12.6g,450.0mmol),剧烈搅拌1小时后,加入饱和的氯化铵溶液淬灭反应,用乙酸乙酯萃取(3×400ml),有机相用饱和氯化钠溶液洗涤、无水硫酸钠干燥,过滤后真空浓缩。然后用硅胶柱层析纯化,得到11.5g结构式为
在-78℃的条件下,将化合物a(9.0g,34.1mmol)溶解于液氨(500ml)中,分批次投入剪碎的钠块(11.8g,511.5mmol),搅拌2小时,得到包括结构式为
小心缓慢的往第二反应液中滴加氯化铵溶液(250ml),淬灭反应后多余的液氨在真空下除去,加入适量盐酸,调节溶液ph=1~2,所得的反应液用乙酸乙酯(3×300ml)萃取,有机相用饱和氯化钠溶液洗涤、无水硫酸钠干燥,过滤后真空浓缩。浓缩产物溶解于甲苯中(100ml),加入对甲苯磺酸(1.44g,10.2mmol),于100℃搅拌2小时,冷却到室温后小心的加入饱和碳酸氢钠溶液(100ml),用乙酸乙酯萃取(3×300ml),有机相用饱和氯化钠溶液洗涤、无水硫酸钠干燥,过滤后真空浓缩。然后用硅胶柱层析纯化,得到3.53g结构式为
将化合物d(1.0g,3.97mmol)溶解于四氢呋喃(30ml)中,依次加入碘(1.51g,5.95mmol)、三苯基膦(1.35g,5.16mmol)、咪唑(405mg,5.95mmol),室温搅拌3小时,加入饱和氯化铵溶液(100ml)淬灭反应,用乙酸乙酯萃取(3×300ml),有机相用饱和氯化钠溶液洗涤、无水硫酸钠干燥,过滤后真空浓缩。然后用硅胶柱层析纯化,得到1.44g结构式为
将化合物d1(2.1g,5.80mmol)溶解于甲苯(50ml)中,加入1,8-二氮杂二环十一碳-7-烯(8.6ml,58.0mmol),在80℃搅拌反应3小时,加入饱和氯化铵溶液(100ml)淬灭反应,用乙酸乙酯萃取(3×300ml),有机相用饱和氯化钠溶液洗涤、无水硫酸钠干燥,过滤后真空浓缩。然后用硅胶柱层析纯化,得到0.68g牛樟芝素(产率50%)。牛樟芝素的检测数据如下:
实施例2
在无水的条件下,将结构式为
在第一反应液中加入硼氢化钠(12.6g,450.0mmol),剧烈搅拌1小时后,加入饱和的氯化铵溶液淬灭反应,用乙酸乙酯萃取(3×400ml),有机相用饱和氯化钠溶液洗涤、无水硫酸钠干燥,过滤后真空浓缩。然后用硅胶柱层析纯化,得到11.5g结构式为
在-78℃的条件下,将化合物a(9.0g,34.1mmol)溶解于液氨(500ml)中,分批次投入剪碎的钠块(11.8g,511.5mmol)后加入乙醇(10ml),搅拌2小时,得到包括结构式为
小心缓慢的往第二反应液中滴加乙醇(100ml),淬灭反应后多余的液氨在真空下除去,加入适量盐酸,调节溶液ph=1~2,所得的反应液用乙酸乙酯(3×300ml)萃取,有机相用饱和氯化钠溶液洗涤、无水硫酸钠干燥,过滤后真空浓缩。浓缩产物溶解于甲苯中(100ml),加入对甲苯磺酸(1.44g,10.2mmol),于60℃搅拌1小时,冷却到室温后小心的加入饱和碳酸氢钠溶液(100ml),用乙酸乙酯萃取(3×300ml),有机相用饱和氯化钠溶液洗涤、无水硫酸钠干燥,过滤后真空浓缩。然后用硅胶柱层析纯化,得到4.76g结构式为
将化合物d(1.0g,3.97mmol)溶解于四氢呋喃(30ml)中,依次加入碘(1.51g,5.95mmol)、三苯基膦(1.35g,5.16mmol)、咪唑(405mg,5.95mmol),室温搅拌3小时,加入饱和氯化铵溶液(100ml)淬灭反应,用乙酸乙酯萃取(3×300ml),有机相用饱和氯化钠溶液洗涤、无水硫酸钠干燥,过滤后真空浓缩。然后用硅胶柱层析纯化,得到1.44g结构式为
将化合物d1(2.1g,5.80mmol)溶解于甲苯(50ml)中,加入1,8-二氮杂二环十一碳-7-烯(8.6ml,58.0mmol),在80℃搅拌反应3小时,加入饱和氯化铵溶液(100ml)淬灭反应,用乙酸乙酯萃取(3×300ml),有机相用饱和氯化钠溶液洗涤、无水硫酸钠干燥,过滤后真空浓缩。然后用硅胶柱层析纯化,得到0.68g牛樟芝素(产率50%)。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!