新型的天冬氨酸衍生物,它的制备方法及其在固相多肽合成中的用途与流程
本方法涉及天冬氨酸的修饰,抑制天冬酰亚胺形成的方法。
背景技术:
蛋白质的化学合成作为当今世界重要的科学前沿领域,很多国家在这个领域投入了大量的人力物力财力。而我国在蛋白质化学合成的领域工作还比较少。本项研究将从蛋白质的合成方法入手,开展相关研究。
基于Fmoc保护的固相多肽合成技术是目前应用比较广泛的合成多肽的方法。当多肽合成仪出现以后,大大提高了合成多肽的效率。但是,固相多肽合成技术还有许多制约其应用的缺陷,比如在碱性条件下去除Fmoc保护基的时候会产生副产物天冬酰亚胺(aspartimide)。
如机理1所示,在碱性条件下,肽链中β羧基位叔丁基保护的天冬氨酸,其临近的氨基酸为甘氨酸、天冬氨酸、天冬酰胺、蛋氨酸、精氨酸时容易生成含有天冬酰亚胺的副产物。这种副产物极易消旋,且在后处理过程中可能进一步开环生成消旋的多肽或异构的β多肽,以及相应的哌啶衍生物。在合成较长多肽时,这一副反应愈加明显。随着合成多肽序列的增长,这些副产物不仅大大降低了目标多肽产物的产率,更因为副产物与目标产物相似的理化性质,极大的增加了分离的难度和成本。通常情况下,在长度大于30个氨基酸的多肽序列中混入相应的天冬亚酰胺副产物,用液相色谱难以纯化出相应的目标多肽产物。
为了解决这一问题,研究者们提出了一系列的解决方案:包括保护α氨基、加入添加剂中和去除Fmoc保护基必须的碱性条件以及增大β羧酸保护基体积等。这些方法对某些多肽的合成取得了良好的效果,但是同时也存在着脱保护基困难、添加剂价格昂贵,且脱Fmoc效率低下、相应天冬氨酸衍生物制备困难且易分解,效果受氨基酸序列影响较大等局限性。找到抑制天冬酰亚胺副产物生成的更好方案,是高效高产率制备出含更多氨基酸的多肽片断和蛋白质、并进行更深入研究所必须的物质保障。为了蛋白质合成的进一步研究,我们将从抑制天冬酰亚胺生成的角度研究提高目标多肽序列产率和纯度的解决方案。
技术实现要素:
本发明在天冬氨酸的β位引入一个较大体积的巯基基团来阻止天冬酰亚胺的形成。本发明的解决方案从分子构象对化学反应活性的影响这一角度出发,设计出β位含巯基取代的天冬氨酸衍生物,通过位阻及分子构象抑制天冬酰亚胺的形成。
本发明的结构式
缩写为
本发明的机理如机理2所示,从易得的天冬氨酸衍生物a出发合成得到化合物b(R为体积较大的基团)。衍生物b在多肽合成中缩合得到残基c,在后续反应条件中若要形成天冬酰亚胺,需经过位阻极大的构象A,这一副反应将被大大抑制。考虑到取代基可能对肽键形成造成的影响,我们将选择不同的巯基保护基并从中筛选出效果最好的衍生物。
得到化合物b后,本发明将选择一系列易形成天冬酰亚胺的序列,例如H-Lys-Asp-Gly-Tyr-Ile-OH,,充分评估抑制天冬酰亚胺形成的效果。
附图说明
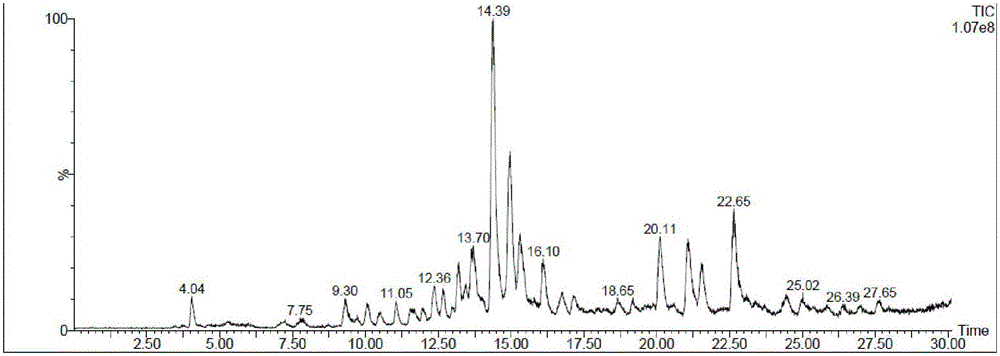
图1是Fmoc-Lys-Xaa-Gly-Tyr-Ile-OH(Xaa是化合物1)LC-MS的紫外吸收图,
图2是Fmoc-Lys-Xaa-Gly-Tyr-Ile-OH(Xaa是化合物1)LC-MS的丰度图,
图3是Fmoc-Lys-Xaa-Gly-Tyr-Ile-OH(Xaa是化合物1)LC-MS22.65分钟的MS图,其中938.15是化合物8的分子量,
图4是Fmoc-Lys-Xaa-Gly-Tyr-Ile-OH(Xaa是化合物1)LC-MS25.02分钟的MS图,其中920.12是天冬酰亚胺的分子量,
实施例
(1)化合物5的合成:将化合物1(1g,3mmol)溶于N,N-二甲基甲酰胺(10ml)中,加入N,N-二异丙基乙胺(0.8ml,4.5mmol)和丙烯基溴(0.34ml,4.44mmol)。室温反应搅拌过夜,用薄层层析监测反应,待反应完成后加入酸终止反应。加入乙酸乙酯稀释反应,分别用水和饱和食盐水洗涤有机层,用无水硫酸镁干燥有机相,抽滤,旋干后过硅胶柱,洗脱剂为石油醚:乙酸乙酯(9:1),最终得到无色油状,产率95%。1H NMR(400MHz,CDCl3)δ5.87(dddd,J=5.3,5.6,10.2,16.9Hz,1H),5.48(br d,J=8.4Hz,1H),5.31(dq,J=16.8,1.5Hz,1H),5.22(dq,J=10.5,1.1Hz,1H),4.65(ddt,J=13.0,5.5,1.5Hz,1H),4.63(ddt,J=13.4,
5.7,1.5Hz,1H),4.54(dt,J=4.5,8.8Hz,1H),2.92(dd,J=4.6,17.1Hz,1H),2.91(dd,J=4.6,16.9Hz,1H),1.46(s,9H),1.45(s,9H)ppm;13C NMR(100MHz,CDCl3)δ171.1,170.2,155.6,131.7,118.6,81.7,80.1,66.2,50.3,38.0,28.4,28.1ppm;
(2)化合物3的合成:将化合物2(0.26ml,1.33mmol)溶于乙醚中,将反应体系降到0℃,滴加磺酰氯(0.11ml,1.33mmol)。在0℃反应10分钟后,将溶于丙酮的4-甲基-硫代苯磺酸钾盐(617.7mg,2.66mmol)滴加于反应体系中。逐步升温至室温,并反应45分钟。用薄层层析监测反应,待反应完成后加入乙酸乙酯稀释反应,分别用水和饱和食盐水洗涤有机层,用无水硫酸镁干燥有机相,抽滤,旋干后过硅胶柱,洗脱剂为石油醚:乙酸乙酯(14:1),最终得到无色油状,产率76%。1H NMR,13C NMR来自DOI:10.1021/jacs.6b01202。
(3)化合物6的合成:将化合物5(520.8mg,1.58mmol)溶于超干四氢呋喃中,氩气保护,并将温度降至-78℃。滴加LiHMDS(5.16mmol)并反应1小时。将溶于超干四氢呋喃的化合物3(437.2mg,1.58mmol)滴加在反应溶液中并反应2小时。用薄层层析监测反应,待反应完成后加入1M盐酸终止反应。加入乙酸乙酯稀释反应,分别用1M盐酸、水和饱和食盐水洗涤有机层,用无水硫酸镁干燥有机相,抽滤,旋干后过硅胶柱,洗脱剂为石油醚:乙酸乙酯(9:1),最终得到黄色油状,产率71%。1H NMR(400MHz,CDCl3)δ6.00–5.81(m,1H),5.67(d,J=9.6Hz,1H),5.38(d,J=16.4Hz,1H),5.32–5.20(m,1H),,4.96–4.78(m,1H),4.65(qt,J=18.0,8.9Hz,2H),3.94(d,J=4.3Hz,1H),1.47(s,9H),1.43(s,9H),1.32(s,9H);13C NMR(101MHz,CDCl3)δ169.84,169.48,154.96,131.47,118.77,83.11,81.59,66.33,54.83,54.48,48.41,29.75(三碳重叠),28.26(三碳重叠),27.90(三碳重叠)。
(4)化合物7的合成:将化合物6(313.3mg,0.58mmol)和四三苯基膦钯(33.4mg,0.029mmol)溶于超干四氢呋喃中,氩气保护,加入N-甲基苯胺(0.13ml,1.16mmol)。室温反应。。用薄层层析监测反应,待反应完成后直接旋干并过硅胶柱,洗脱剂为石油醚:乙酸乙酯(3:1,1%AcOH),最终得到黄色油状,产率76%。1H NMR(400MHz,CDCl3)δ5.81(5.39)*(d,J=8.5Hz,1H),4.86(4.79)*(d,J=5.7Hz,1H),3.98(3.93)*(d,J=3.2Hz,1H),1.47(s,9H),1.41(s,9H),1.36(s,9H).13C NMR(101MHz,CDCl3)δ174.31(173.85)*,169.86(168.10)*,156.05,83.91(83.11)*,80.64,59.01(57.41)*,55.03(54.37)*,48.57,29.75,28.24,27.88.带有“*”表示有一对旋转异构体。
(5)化合物1的合成:将化合物7(153.6mg,0.29mmol)溶于二氧六环(含4MHCl)中,并加入三乙基硅烷(0.46ml,2.9mmol),室温反应4小时。用氩气将二氧六环(含4MHCl)和三乙基硅烷吹干。用二氯甲烷溶解中间产物。加入Fmoc-OSu(391.3mg,1.16mmol)和三乙胺(0.16ml,1.16mmol),室温反应过夜。用薄层层析监测反应,待反应完成后加入1M盐酸终止反应。加入乙酸乙酯稀释反应,分别用1M盐酸、水和饱和食盐水洗涤有机层,用无水硫酸镁干燥有机相,抽滤,旋干后过硅胶柱,洗脱剂为石油醚:乙酸乙酯(1:1,1%AcOH),最终得到黄色油状,产率63%。1H NMR(400MHz,CDCl3)δ7.75(d,J=12.9Hz,2H),7.63(d,J=7.3Hz,2H),7.40(d,J=7.3Hz,2H),7.31(d,J=14.1Hz,2H),6.10(d,J=9.3Hz,1H),5.04(d,J=6.5Hz,1H),4.42(d,J=7.2Hz,2H),4.08(s,1H),1.50(s,,9H),1.35(s,9H).13C NMR(101MHz,CDCl3)δ174.48,169.37,156.51,143.73(两碳重叠),141.24(两碳重叠),129.00(两碳重叠),128.19(两碳重叠),127.67(两碳重叠),125.24(两碳重叠),84.05,67.64,57.46,54.57,48.72,47.04,29.77,27.88。
(6)化合物8的合成:通过多肽固相合成法(solid phase peptide synthesis简写为SPPS)合成Fmoc-Lys-Xaa-Gly-Tyr-Ile-OH(Xaa是化合物1)。
根据LC-MS,图1是LC-MS的紫外吸收图,图2是LC-MS的丰度图,图3是22.65分钟的MS图,其中938.15是化合物11的分子量,图4是25.02分钟的MS图,其中920.12是天冬酰亚胺的分子量,根据化合物的丰度,可以断定此方法部分有效的抑制了天冬酰亚胺的形成。
- 还没有人留言评论。精彩留言会获得点赞!