选择性IL‑6‑跨信号转导抑制剂的给予的利记博彩app
相关申请的引证
本申请要求2014年12月1日提交的美国临时申请号62/086,054的权益,其内容在此通过引证以其整体并入。
背景技术:
il-6是由造血细胞和非造血细胞产生的多效细胞因子,例如响应于感染和组织损伤。il-6通过两种主要信号转导途径(signallingpathway,信号转导通路,信号途径,信令途径)发挥其多重生物活性,一种是通过主要存在于肝细胞和某些白细胞上的膜结合il-6r的所谓经典配体-受体途径,以及一种是通过源自膜结合il-6r的蛋白水解切割或来自选择性剪接的循环sil-6r的跨信号转导途径(trans-signallingpathway,反式信号途径)。
在经典途径中,il-6直接与有限范围细胞类型的表面上的膜结合的il-6r结合。il-6/il-6r复合物与信号转导性gp130受体蛋白质的预先形成的二聚体缔合,引起gp130同二聚体的空间变化,并从而引发细胞内信号转导级联(signallingcascade)。经典信号转导负责急性炎症防御机制和关键的生理学il-6功能,如肠上皮细胞的生长和再生信号。
il-6r和gp130的细胞外结构域可通过翻译选择性剪接的mrna而产生,无膜-锚定结构域,从而产生sil-6r和sgp130变体。另外,il-6r的细胞外结构域可以通过a去整合蛋白和金属蛋白酶(adam)家族(在人类中,adam17)的膜结合蛋白酶而脱落,以产生sil-6r。在跨信号转导过程中,sil-6r结合至il-6,形成激动复合物,其结合存在于不表达膜结合il-6r的许多细胞类型上的跨膜gp130二聚体;然后在通常不响应il-6的细胞中诱导通过信号转导物和转录激活因子(stat)的il-6信号转导。il-6/sil-6r复合物的活性通常由循环中存在的sgp130的水平控制,其与膜结合的gp130有效竞争。跨信号转导主要参与慢性炎症,并且已被证明可预防促炎粘膜t细胞群体进入细胞凋亡。
希望具有模拟天然跨信号转导抑制剂sgp130但具有更高结合亲和力并因此具有更强的抑制活性的分子。此外,期望具有能以最小毒性和免疫原性潜力向人施用的分子。
技术实现要素:
现在已经发现,选择性il-6-跨信号转导抑制剂可以在大剂量范围内施用于人类而没有任何显著的有害作用。此外,令人惊讶地发现,该抑制剂的末期半衰期允许每周一次、每两周(即每隔一周)、每月或甚至更少的频率的剂量。
在某些实施方式中,本发明包括用于治疗炎性疾病或il-6介导的病症的抑制剂(例如,如本文所公开的多肽二聚体),其中多肽以0.5mg至5g的剂量给予。本发明还包括通过给予抑制剂(例如,本文所公开的多肽二聚体)以治疗炎性疾病的方法,其中抑制剂的剂量为0.5mg至5g。本发明进一步包括此种抑制剂在制造用于以指定的剂量治疗炎性疾病的药物中的用途。优选地,治疗人类。
在其它实施方式中,本发明包括本文公开的多肽二聚体,其用于治疗il-6介导的病症而不会显著降低中性粒细胞(中性白细胞)计数、血小板计数和/或c反应蛋白的水平,或者不会使健康受试者或患有il-6介导的病症的患者中的中性粒细胞计数、血小板计数和/或c反应蛋白的水平降低到低于正常范围。本发明还包括通过给予本文公开的多肽二聚体而治疗il-6介导的病症的方法,其中所述方法不会显著地降低中性粒细胞计数、血小板计数和/或c反应蛋白的水平。本发明进一步包括此种多肽二聚体在制造用于治疗il-6介导的病症而不会显著降低中性粒细胞计数、血小板计数和/或c反应蛋白的水平的药物中的用途。优选地,治疗人类。
附图说明
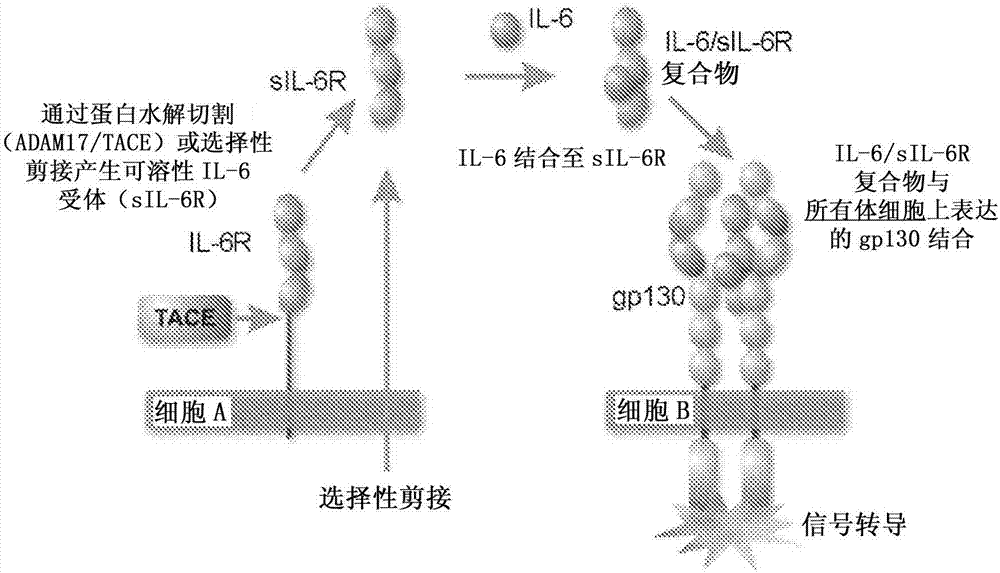
图1示出了il-6的跨信号转导途径。从选择性剪接的mrna或蛋白水解切割产生的sil-6r能够结合il-6以形成il-6/sil-6复合物,该复合物结合存在于绝大多数体细胞类型上的gp130并诱导细胞内信号转导级联。
图2显示,包含两个seqidno:1的单体的多肽二聚体不干涉结合至膜结合的il-6r的il-6(经典信号转导),但选择性地结合il-6/sil-6r复合物并且防止跨信号转导。
图3示出了以0.75mg、7.5mg、75mg、150mg、300mg、600mg和750mg静脉注射输注肽1后的分布图(左图)以及以60mg(2x2ml)皮下注射肽1后的分布图(右图)。
图4示出了以75mg、300mg和750mg,在健康受试者中静脉给予后的分布图(左图)和在临床缓解的cd患者中静脉给予后的分布图(右图)。
图5示出了在健康受试者中以75mg、300mg和750mg每周一次连续4周静脉给予后的分布图。
图6示出了使用2室结构pk模型的模型预测(实线)以及试验000115中的观察数据(圆圈)。
图7示出了单个gp130-fc亚单元的核苷酸和氨基酸序列。
图8示出了表达质粒pfer02的核苷酸序列元素。
具体实施方式
本发明的优选抑制剂包括两个gp130-fc融合单体(例如两个seqidno:1的单体)的二聚体。在其活性形式时,seqidno:1的多肽作为由处于cys623和cys626的两个二硫键连接的二聚体存在(图2)。seqidno:2对应于具有内源信号肽的gp130-fc融合单体的氨基酸序列。在蛋白质合成期间除去信号肽,导致产生seqidno:1的多肽。
本文所述的多肽二聚体选择性地抑制过多的跨信号转导(图1)并且诱导参与多发性炎性疾病的有害t细胞的凋亡。多肽二聚体靶向并中和il-6/sil-6r复合物,并因此预期仅在所需治疗浓度下抑制il-6跨信号转导,保持经典信号转导及其许多生理功能以及其急性炎症防御机制完整无缺(图2)。认为,多肽二聚体由于位阻而不能干扰经典il-6信号转导;fc部分不能插入细胞膜,使得gp130部分不能结合到膜结合的il-6/sil-6r复合物。因此,预期多肽二聚体具有与全局il-6阻断(例如托西珠单抗、sirukumab)相似的功效,但其中副作用更少。
本文所述的多肽二聚体优选包含具有对应于seqidno:1的序列的gp130-fc单体。在某些实施方式中,单体具有对应于seqidno:2的序列。在某些实施方式中,本文描述的多肽二聚体包含与seqidno:1或seqidno:2具有至少90%、95%、97%、98%、99%或99.5%序列同一性的多肽。优选地,多肽包含gp130d6结构域(特别是氨基酸tfttpkfaqge:seqidno:1的氨基酸位置585-595)、fc结构域铰链区中的aega(seqidno:1的氨基酸位置609-612)并且在gp130部分和fc结构域之间不包含接头(linker)。在优选的实施方式中,本公开提供了多肽二聚体,其包含两个与seqidno:1具有至少90%序列同一性的氨基酸序列的单体,其中氨基酸序列包含gp130d6结构域、fc结构域铰链区中的aega并且在gp130部分和fc结构域之间不具有接头。在优选的实施方式中,本公开提供了多肽二聚体,其包含两个与seqidno:2具有至少90%序列同一性的氨基酸序列的单体,其中氨基酸序列包含gp130d6结构域、fc结构域铰链区中的aega并且在gp130部分和fc结构域之间不具有接头。
期望的是,多肽基本上不含半乳糖-α-1,3-半乳糖部分,因为这些与免疫原性反应相关。令人惊讶地发现,本发明的二聚体具有低水平的这种部分。在优选的实施方式中,多肽二聚体含有每摩尔多肽不大于6%的半乳糖-α-1,3-半乳糖。优选地,多肽二聚体含有不大于4mol%、3mol%、2mol%、1mol%、0.5mol%、0.2mol%、0.1mol%或甚至不可检测水平的半乳糖-α-1,3-半乳糖(例如,通过wax-hplc、np-hplc或wax测量,优选通过wax-hplc测定)。在其它实施方式中,以质量或摩尔计,相对于聚糖的总量,多肽二聚体含有少于6%、4%、3%、2%、1%、0.5%、0.2%或甚至0.1%的半乳糖-α-1,3-半乳糖。
还希望本发明的多肽被唾液酸化。这具有增加本发明多肽的半衰期的优点。多肽二聚体的每个链含有10个n-糖基化位点;9个n-糖基化位点位于gp130部分中,1个n-糖基化位点位于fc部分中。因此,多肽总共含有20个糖基化位点。在某些实施方式中,在多肽上平均至少52%或至少54%的聚糖包括唾液酸残基,诸如平均52-65%(例如通过wax-hplc、np-hplc或wax测量,优选通过wax-hplc测量)。优选地,本发明的多肽具有220kda的近似分子量;每93kda具有源自10个n-糖基化链的额外的~20kda的分子量。
进一步期望使多肽聚集的程度最小化,这在本文中称为低聚,产生低聚聚集体。如本文中使用的“低聚聚集体”不是指活性二聚肽。相反,该术语是指至少活性二聚体的二聚体。令人惊讶地发现,本发明的肽二聚体显示低水平的聚集。在某些实施方式中,低于5%、低于4%、低于3%、低于2%、低于1.5%或甚至低于1.0%的多肽以低聚物存在。低聚物含量可以通过例如尺寸排阻色谱-多角度光散射(sec-mals)或sec-uv来测量。
优选地,多肽二聚体以其全长形式存在(例如包括两个全长的单体,例如seqidno:1的单体)。然而,细胞培养可以产生本文称为单个gp130形式(sgf)的截短变体。sgf是共价结合的双链分子,一条链包含全长gp130-fc单体(例如,seqidno:1的单体)并且第二链包含截短的gp130-fc单体(例如,截短的seqidno:1),该第二链包括fc结构域并且缺少大部分或全部的gp130结构域(例如,在fc区的接头序列之前终止)。迄今为止的研究表明sgf不具有异质氨基末端。sgf可以在生物反应器中以一致的水平形成,并且一旦形成,sgf水平在纯化、加工或加速储存条件下不容易改变。由于与多肽二聚体的全长形式相似的物理-化学性质,sgf水平在纯化过程中难以除去;因此去除sgf的努力可能导致产量的显著降低。令人惊讶地发现,本发明的多肽二聚体几乎总是全长的。在某些实施方式中,本发明的组合物包含的多肽二聚体包含不大于4.0重量%、3.0重量%、2.0重量%或甚至1.5重量%的多肽,该多肽相对于seqidno:1的多肽是seqidno:1的多肽的截短变体。在某些实施方式中,本发明的组合物包含不大于4.0重量%、3.0重量%、2.0重量%或甚至1.5重量%的多肽,该多肽相对于seqidno:2的多肽是seqidno:2的多肽的截短变体。
剂量给药
基于i期数据,本文所述的剂量代表了被认为是安全且可耐受的剂量范围。在早期临床试验中,在健康受试者和ra患者中,靶向il-6r或il-6的其他化合物经常显示出降低的中性粒细胞计数和血小板计数以及降低c反应蛋白的水平(crp)。然而,在使用本发明的多肽剂量给药的健康受试者中,观察到的中性粒细胞计数和血小板计数的水平仍处于正常范围内。从i期程序的结果来看,本发明的多肽没有显示出与靶向il-6r或il-6的化合物对生物标志物相同的作用。
在包含seqidno:1的单体的多肽二聚体的两个试验中,采用通过刺激来自具有超il-6的受试者的全血样品来测量stat3的活化水平的离体测定,作为药物活性的评估。据信,高于1μg/ml的包含seqidno:1的单体的多肽的浓度水平,涉及第二信使(stat3)测定中将信号抑制至基线。包含seqidno:1的单体的多肽二聚体的浓度水平对应于7.5mg剂量的峰水平。作为静脉输注给药的75mg剂量,已显示在一周给药间隔的稳定状态下具有高于1μg/ml的浓度。每两周给药的相应剂量为300mg。据信,作为皮下注射给药的60mg被认为导致了每周剂量给药的相同稳定状态。
在某些实施方式中,剂量为0.5mg至5g多肽二聚体。例如,剂量可以为5mg至3g,10mg至2g,60mg至1g或优选60mg至750mg。
多肽二聚体可以以适合于预期病症的频率给药。在某些实施方式中,多肽二聚体每7-60天给予一次。例如,多肽二聚体可以每7-30天或7-20天给予一次。在优选的实施方式中,给予每周一次(每7天一次)或每两周一次(每14天一次)。给予也可以每天进行或每周进行两次或三次。剂量是指单剂量事件,不论剂量是单位剂量形式还是多个单位剂型组合在一起(例如,摄取两个或更多个药丸,接受两次或更多次注射)。如下所讨论的,这种剂量频率不能从动物研究预测。人类临床试验发现平均半衰期为4.6天至5.5天。相比之下,当静脉内给予多肽二聚体时,食蟹猴具有的半衰期仅为0.7天,并且皮下给予时的半衰期仅为1.4-1.5天。
本发明的多肽二聚体通常肠胃外给予,如静脉或皮下给予。可以根据本文公开的剂量给予频率之一进行给予。
在某些实施方式中,静脉给予多肽二聚体,每7-60天给予一次,剂量为60mg至1g。
在某些此种实施方式中,静脉给予多肽二聚体,每7-30天给予一次,剂量为60mg至1g。
在示例性的实施方式中,静脉给予多肽二聚体,每周给予一次,剂量为60mg至1g。
在另一示例性实施方式中,静脉给予多肽二聚体,每两周给予一次,剂量为60mg至1g。
在某些实施方式中,皮下给予多肽二聚体,每7-60天给予一次,剂量为60mg至600g。
在某些实施方式中,皮下给予多肽二聚体,每7-30天给予一次,剂量为60mg至600g。
在示例性实施方式中,皮下给予多肽二聚体,每周给予一次,剂量为60mg至600g。
在另一示例性实施方式中,皮下给予多肽二聚体,每两周给予一次,剂量为60mg至600g。
安全性
包含seqidno:1的单体的多肽二聚体已经作为单剂量给予高达750mg以及连续4周每周一次给予600mg。多肽的安全性良好,在包括安慰剂组在内的所有治疗组中发生的不良事件很少,且均为轻度或中度。没有观察到不良事件的发生率或频率的明显剂量相关趋势。生命指征、ecg或临床化学参数没有明显的剂量相关趋势或治疗相关的变化。发生三次输液反应事件,均为轻度/中度,伴随有皮肤症状如荨麻疹和肿胀,并且迅速消退,无任何后遗症。
总体上,当连续4周每周一次静脉注射给予高达600mg以及作为单次剂量静脉注射给予高达750mg时,包含seqidno:1的单体的多肽二聚体是安全的并且耐受性良好。
通过分析研究靶向il-6r或il-6的类似化合物的临床研究,也可以间接地解决包含seqidno:1的单体的多肽在人体中的潜在风险。迄今,没有与该多肽二聚体相同的方式阻断信号转导途径的批准的化合物,即靶向并中和il-6/sil-6r-复合物以抑制跨信号转导途径,而不与单独的il-6或il-6r相互作用。然而,有靶向il-6受体的化合物的经验。这些化合物之一是托西珠单抗,其已在欧洲和美国获得批准。托西珠单抗特异性结合可溶性和膜结合的il-6受体,并已显示抑制sil-6r和mil-6r介导的信号转导。
使用托西珠单抗治疗的ra患者中所报道的最常见的不良药物反应(发生≥5%)是上呼吸道感染、鼻咽炎、头痛、高血压和alt升高。最严重的不良药物反应是严重感染、憩室炎并发症和超敏反应。用托西珠单抗治疗后发生中性粒细胞减少和血小板计数减少。在托西珠单抗8mg/kg加改良抗风湿药物(dmard)治疗的患者中有3.4%发生了中性粒细胞计数降低至低于109/l。大约一半发展有anc<109/l的患者在开始治疗后的8周内是这样的。在0.3%接收托西珠单抗8mg/kg和dmard的患者中报道了低于5x108/l的降低。严重的中性粒细胞减少可能与严重感染的风险增加相关,尽管迄今为止在托西珠单抗的临床试验中,中性粒细胞减少和严重感染发生之间没有明显的关联。输注期间报告的事件主要是高血压发作;完成输注后24小时内报告的事件为头痛和皮肤反应(皮疹,荨麻疹)。这些事件不是治疗限制的。在受控和开放标签临床研究期间,用托西珠单抗治疗的3,778例患者中,共有13例(0.3%)报道了与托西珠单抗相关的临床显著超敏反应并且需要停止治疗。这些反应通常在托西珠单抗的第二至第五次输注期间被观察到的。在托西珠单抗临床试验和上市后,报道了罕见事件的胃肠穿孔,主要在具有憩室炎史的患者中。病因尚不清楚,但是ra患者通常增加了上下gi肠穿孔的风险(与dmard疗法无关);在糖皮质激素治疗的ra患者、nsaid或具有憩室炎史的ra患者中风险最高。在托西珠单抗治疗期间,营销授权后已报道了致命性过敏反应。应注意的是,ra患者可能有其他背景疾病作为混杂因素。
由于包含seqidno:1的单体的多肽二聚体是一流的融合蛋白,因此,与具有不同作用机制的不同单克隆抗体产品的比较是价值有限的。与其他完全阻断il-6活性的产品相反,这种多肽二聚体被认为只会干扰il-6/sil-6r复合物,使膜结合的il-6途径可接近。
il-6广泛地参与机体免疫和炎症反应。当可溶性和膜结合的il-6r都被阻断时,潜在的感染和其他免疫依赖性疾病的风险增强且炎症反应较不突出。虽然不希望被理论束缚,但据信,用仅靶向il-6/sil-6r复合物的包含seqidno:1的单体的多肽二聚体的治疗,将阻止慢性肠道炎症在ibd中的持续并保留由经典il-6信号转导激活的急性期炎症反应,从而降低机会性感染的风险。然而,不能预测是否存在本发明的多肽二聚体的浓度,在这种浓度下经典il-6信号转导将受到影响。因此,本文的数据令人惊奇地证明,相对于靶向il-6活性的其他治疗,对经典il-6信号转导的影响较小。
基于本文提供的数据,本发明的多肽二聚体的优点是,与抑制il-6的其他化合物相比,它可能对中性粒细胞计数、血小板计数和/或c反应蛋白的水平影响较小。在某些实施方式中,本发明的多肽二聚体不会显著降低中性粒细胞计数、血小板计数和/或c反应蛋白的水平,或者不会在健康受试者或者患有il-6介导的病症的患者中使中性粒细胞计数、血小板计数和/或c反应蛋白的水平降低至低于正常范围。例如,以本文描述的剂量给予多肽二聚体将中性粒细胞计数、血小板计数和/或c反应蛋白的水平维持在正常生理范围内。在某些实施方式中,中性粒细胞计数、血小板计数和/或c反应蛋白的水平低于正常生理范围下限不超过50%、40%、30%、20%、15%、10%或5%。中性粒细胞计数、血小板计数和/或c反应蛋白的水平的测量可以在治疗后立即、治疗后1天、治疗后3天、治疗后1周、治疗后2周、治疗后1个月、治疗后3个月、治疗后6个月或治疗后一年进行。
中性粒细胞计数、血小板计数和c反应蛋白的水平的测定可以通过本领域熟知的任何数量的测定进行。中性粒细胞计数,也称为绝对中性粒细胞计数(anc),是血液中存在的中性粒细胞数量的量度(参见例如al-gwaizla,babayhh(2007).“thediagnosticvalueofabsoluteneutrophilcount,bandcountandmorphologicchangesofneutrophilsinpredictingbacterialinfections”.medprincpract16(5):344–7)。成年男性和女性的c-反应蛋白的正常生理值为0-5.00mg/l(例如,通过比浊法)。成年女性的中性粒细胞的正常生理值为1.61-6.45x109/l(绝对值,例如通过激光流式细胞术)或37.9-70.5%(计算的);在成年男性中,相应值为1.46-5.85x109/l和38.2-71.5%。成年女性的血小板的正常生理值为173-369x109/l(例如通过高频阻抗测量);在成年男性中,相应的值是155-342x109/l。
本发明的多肽二聚体优选在人体中不显著诱导抗体的形成(例如,针对多肽二聚体的抗体)。甚至更优选地,抗体不是中和抗体。在某些实施方式中,在低于5%、2%、1%、0.5%、0.2%、0.1%或0.01%的经治疗的受试者或患者中可检测到针对本发明的多肽二聚体的抗体。典型地,检测限约为9ng/ml血清。
指征
在急性炎症中,已经显示il-6在肝脏中诱导急性期反应,导致急性期蛋白级联的释放,特别是crp。通过在炎症部位形成由细胞凋亡中性粒细胞脱落的sil-6r的复合物,并将得到的il-6/sil-6r跨信号转导复合物与内皮细胞上的信号转导体gp130结合,il-6诱导趋化因子如单核细胞趋化蛋白(mcp)-1的表达,并吸引单核细胞。这导致急性炎症的消退和适应性免疫应答的开始。因此,在急性炎症中,il-6与sil-6r的复合物支持早期主要是中性粒细胞炎症阶段和更持久的单核细胞流入之间的转变,最终也导致炎症的消退。
慢性炎症,如克罗恩氏病(cd)、溃疡性结肠炎(uc)、类风湿性关节炎(ra)或牛皮癣,在组织学上与单核细胞如巨噬细胞和淋巴细胞的存在有关,在获得用于解决急性炎症期后持续存在于组织中。在慢性炎性疾病模型中,通过在t细胞上诱导持续的mcp-1分泌、血管增生和抗凋亡功能,il-6似乎具有有利于损伤部位的单核细胞积累的有害作用。
炎症性肠病(ibd),即cd或uc,是易感个体肠道中发生的慢性炎症,其被认为与特定病原体无关。上皮粘膜屏障中的改变伴随着肠道通透性增加导致粘膜免疫系统增强暴露于肠腔抗原,这导致患者肠道免疫系统的不适当活化。粘膜cd4+t淋巴细胞的不受控制的激活伴随着促炎细胞因子的连续过度释放,诱导致病性胃肠道炎症和组织损伤。有一个共识,即参与ibd发病机制的主要活化免疫细胞是肠t细胞和巨噬细胞。
il-6在人体中显示为ibd中的中枢细胞因子。与对照组相比,已经发现cd和uc患者产生升高水平的il-6,il-6水平与临床活性相关。也发现cd患者的sil-6r水平升高,并因此血清中il-6/sil-6r复合物的水平升高。从cd和uc患者的手术结肠标本获得的固有层固有单核细胞显示,与对照相比,cd4+t细胞和巨噬细胞都产生增加的il-6量。发现sil-6r通过从巨噬细胞和单核细胞的表面脱落而释放,伴随着与il-6水平升高相关的生产增加。在cd患者中,粘膜t细胞显示出对il-6跨信号转导的强烈证据,伴随有stat3、bcl-2和bcl-xl的活化。il-6跨信号转导的阻断引起t细胞凋亡,表明il-6/sil-6r系统在cd中介导t细胞对细胞凋亡的抗性。
因此,在ibd患者中,导致炎症持续存在的固有层中的促炎cd4+t细胞的获得性积累严重依赖于抗凋亡il-6/sil-6r跨信号转导。据信,通过作用于il-6/sil-6r复合物,本文公开的多肽二聚体可用于治疗cd和其它炎性疾病。
因此,本发明的多肽二聚体可以治疗il-6介导的病症。il-6介导的病症包括炎性疾病或癌症。在这方面,本文所述的多肽和组合物可以施用于患有炎性疾病的受试者,如幼年特发性关节炎、克罗恩氏病、结肠炎(例如与ibd无关的结肠炎,包括放射性结肠炎、憩室结肠炎、缺血性结肠炎、感染性结肠炎、脂泻病、自身免疫性结肠炎或由影响结肠的过敏引起的结肠炎)、皮炎、牛皮癣、葡萄膜炎、憩室炎、肝炎,肠易激综合征(ibs)、红斑狼疮、肾炎、帕金森病、溃疡性结肠炎、多发性硬化症(ms)、阿尔茨海默病、关节炎、类风湿性关节炎、哮喘和各种心血管疾病如动脉粥样硬化和血管炎。在某些实施方式中,炎性疾病选自于由糖尿病、痛风、冷冻蛋白相关周期性综合征和慢性阻塞性肺疾病组成的组。
优选地,炎性疾病或il-6介导的病症是炎症性肠病,优选其中治疗诱导炎症性肠病的缓解。优选地,炎症性肠病是克罗恩氏病或溃疡性结肠炎,优选其中治疗维持炎性肠病的缓解。优选地,炎性疾病或il-6介导的病症是类风湿性关节炎、牛皮癣、葡萄膜炎或动脉粥样硬化。优选地,炎性疾病或il-6介导的病症是与炎症性肠病无关的结肠炎,优选其中结肠炎是放射性结肠炎、憩室结肠炎、缺血性结肠炎、感染性结肠炎、脂泻病、自身免疫性结肠炎或由影响结肠的过敏引起的结肠炎。
对于诸如炎症性肠病的炎性疾病,治疗可以包括病症的缓解、病症缓解的维持或两者。
其他实施方式提供了一种治疗癌症、降低癌症的严重性或预防癌症的方法,所述癌症包括但不限于多发性骨髓瘤、浆细胞白血病、肾细胞癌、卡波西氏肉瘤、结肠直肠癌、胃癌、黑素瘤、白血病、淋巴瘤、神经胶质瘤、多形性成胶质细胞瘤、肺癌(包括但不限于非小细胞肺癌(nsclc;腺癌和鳞状细胞癌))、非霍奇金淋巴瘤、霍奇金病、浆细胞瘤、肉瘤、胸腺瘤、乳腺癌、前列腺癌、肝细胞癌、膀胱癌、子宫癌、胰腺癌、食管癌、脑癌、头颈癌、卵巢癌、子宫颈癌、睾丸癌、胃癌、食管癌、肝癌、急性淋巴细胞白血病(all)、t-all、急性髓性白血病(aml)、慢性粒性白血病(cml)和慢性淋巴细胞性白血病(cll)、唾液癌或其它癌症。
本公开的另外的实施方式提供了一种治疗疾病、降低疾病的严重性或预防疾病的方法,该疾病选自于由败血症、骨吸收(骨质疏松症)、恶病质、癌症相关疲劳、牛皮癣、全身性青少年特发性关节炎、系统性红斑狼疮(sle)、肾小球膜增生性肾小球肾炎、高丙种球蛋白血症、卡斯尔曼氏病、igm丙种球蛋白病、心脏粘液瘤和自身免疫性胰岛素依赖性糖尿病组成的组。
如本文中所使用的,术语“治疗”、“疗法”和“处理”是指逆转如本文所述的疾病或病症或其一个或多个症状、减轻疾病或病症或其一个或多个症状、延迟疾病或病症或其一个或多个症状的发作或抑制疾病或病症或其一个或多个症状的进展。在一些实施方式中,治疗可以在一个或多个症状发展之后施用。在其它实施方式中,治疗可以在没有症状的情况下施用。例如,治疗可以在症状发作之前施用于易感个体(例如考虑到症状史和/或考虑到遗传或其他易感因素)。症状解除后也可继续治疗,例如预防或延迟复发。
本发明的多肽二聚体可以与第二活性剂一起施用。第二活性剂可以是5-氨基水杨酸、硫唑嘌呤(咪唑硫嘌呤)、5-巯基嘌呤和皮质类固醇中的一种或多种。用于施用5-氨基水杨酸、硫唑嘌呤、5-巯基嘌呤和皮质类固醇的剂量方案是本领域技术人员所熟知的。
例如,可以通过在细胞中表达单体例如包含seqidno:1的单体来生产多肽二聚体。在示例性实施方式中,包含编码seqidno:1或seqidno:2的核酸的载体可以被转染到细胞中。表达载体的设计,包括调控序列的选择,可取决于诸如待转化的宿主细胞的选择、所需蛋白质的表达水平等因素。用于哺乳动物宿主细胞表达的调控序列包括在哺乳动物细胞中引导高水平蛋白质表达的病毒元素,如源自逆转录病毒ltr、细胞巨化病毒(cmv)(诸如cmv启动子/增强子)、猿猴病毒40(sv40)(诸如sv40启动子/增强子)、腺病毒(例如腺病毒主要晚期启动子(admlp))、多瘤和强哺乳动物启动子(如天然免疫球蛋白和肌动蛋白启动子)的启动子和/或增强子。宿主细胞可以是哺乳动物、昆虫、植物、细菌或酵母细胞,优选细胞是哺乳动物细胞,诸如cho细胞。
培养转染的细胞以允许细胞表达所需的蛋白质。然后收集细胞和培养基,并例如通过色谱柱步骤(例如mabselectsure,sp琼脂糖,captoq)纯化多肽二聚体。二聚体也可以用病毒还原/灭活步骤进行浓缩和/或处理。然后,所得到的二聚体可以被用来制备组合物,优选有用于疗法的药物组合物。
范例
实施例1
动物研究
实施例1a
小鼠药代动力学
通过静脉注射(3mg/动物)或皮下注射(0.3、3和30mg/动物),四组54只称重为25-38g的小鼠(27只雄性和27只雌性)接收单剂量的活性二聚形式的seqidno:1的多肽(“肽1”)。
生物利用度为约60%,并且观察到了auc、auct和c最大的表观剂量线性。如对于蛋白质所预期的一样,t最大为8-24小时。从体循环中缓慢清除肽1,清除量为142ml/天/kg。通过消除相(vz)和第一时刻曲线(vss)估计的分布体积分别为397ml/kg和284ml/kg,表明肽1分布在血管床外。末期半衰期范围为1.3-2.3天。
实施例1b
大鼠药代动力学
单剂量给予
在两种不同株的大鼠(spraguedawley(8只大鼠/组)和wistar(24只大鼠/组))静脉和皮下施用后研究了肽1的单剂量pk,显示出稍微不同的结果。清除率(分别为57和93ml/kg/天)和分布体积在spraguedawley大鼠中似乎更低,生物利用度高出2倍,60%相比于wistar大鼠的约30%。皮下给予后观察到的t最大为0.5-1.5天,并且末期半衰期为约2天,范围为1.7-2.7天,在两个给予途径之间仅存在微小差异。
重复剂量给予
静脉给予
大鼠(n=18)接收静脉注射推注10、30和100mg/kg/时刻,每周2次,连续2周。在第一次给予后的c最大和auc的增加似乎是近似剂量线性的。然而,在所有剂量水平下,雄性的暴露量显著高于雌性。对于雄性大鼠,100mg/kg/时机组达到了2100μg/ml的c最大水平,而对于雌性大鼠则为1740μg/ml。auct值为942和642天×μg/ml。
两周后,对于10mg/kg和30mg/kg的剂量组,大鼠中肽1的全身暴露减少。由于本研究第8周在所有动物中证实了抗药物抗体(ada)反应,因此随着时间的推移,预期暴露的降低可归因于ada介导的肽1的清除。
皮下给予
在2周和4周的重复剂量皮下研究中,在第一次给予后的c最大和auc似乎约剂量线性地增加。然而,在最后一次剂量下,观察到显著降低的药物暴露,可能是由于抗体形成引起的;所有测试的动物显示针对肽1的抗体。
实施例1c
食蟹猴药代动力学
单剂量给予
在0.1-100mg/kg皮下注射(n=4)和1.0mg/kg静脉注射(n=4)的剂量下,在雄性和雌性食蟹猴中研究了肽1的单剂量给予的pk。在皮下注射给予后的生物利用度为约60%,t最大为6-24小时。清除率为98ml/天/kg,并且分布体积为约70-90ml/kg。静脉注射给予后的半衰期为0.68天,并且在皮下注射给予后的半衰期为约1.5天。
重复剂量给予
向食蟹猴连续2周每周两次皮下注射给予2(n=4)、10(n=4)、50(n=2)或100(n=2)mg/kg的肽1,以及连续4周每周两次皮下注射给予10(n=4)、30(n=4)或100(n=4)mg/kg的肽1。借助于c最大和auc的暴露近似剂量线性地增加,并且在2周后观察到在较高剂量下第一次和最后一次剂量之间的相似暴露和最大浓度。然而,4周后,与第一次给予相比,最后一次给予的剂量显示较低的药物暴露,可能是由于抗体形成引起的。不同治疗组中首次给予的肽1的平均半衰期范围为1.0-1.8天,最后给予后的半衰期较短。
实施例2
临床试验000067(单剂量)
设计
这是一个单剂量、安慰剂对照、单盲、剂量内随机的平行组剂量递增试验。试验分两部分进行,其中第1部分包括健康受试者,第2部分包括临床缓解中的cd患者。目的是检查安全性和耐受性,并且如果可能,在单次剂量的肽1之后获得药理学作用的迹象。
在第1部分中,包括64名受试者,其中48名(44名男性,4名女性)接收活性剂治疗,16名(全部男性)接收安慰剂。调查了七个剂量,它们是历经30分钟(0.75mg、7.5mg、75mg)或历经1小时(150mg、300mg、600mg和750mg)静脉输注给予的。此外,6名受试者接收皮下注射60mg剂量的肽1并且2名受试者接收皮下注射剂量的安慰剂。肽1以处于25mm组氨酸、200mm蔗糖和0.1mg/ml聚山梨醇酯20中的15mg/ml的浓度给予。
在第2部分中,包含24名患者,其中18名(11名男性,7名女性)接收活性剂治疗(75mg、300mg和750mg),并且6名(4名男性,2名女性)接收安慰剂,全部是通过静脉注射给予的。
结果
静脉注射给予肽1后的pk评测显示,auc和c最大的剂量比例均处于0.75mg至750mg范围内,血浆中的c最大浓度在0.2至170μg/ml的范围内(图3)。清除率为约0.13l/h,平均末期半衰期为约4.5天,且分布体积为约20l,后者指示了一些血管外分布。皮下注射给予60mg肽1显示,在2.3天时的c最大为1.1μg/ml,并且半衰期为5.0天。皮下注射给予肽1后,计算的生物利用度为约50%。没有靶介导的药物处置的指征。
向缓解中的cd患者静脉注射给予75、300和750mg,显示出了与健康受试者非常类似的结果(图4)。auc和c最大是剂量成比例的,c最大浓度为16、76和186μg/ml(健康受试者为16、77和161μg/ml)。清除率为约0.13l/h,平均末期半衰期为约4.6天,且分布体积为约22l。
肽1的安全性良好,在包括安慰剂组在内的所有治疗组中发生的不良事件很少,且均为轻度或中度。没有观察到不良事件的发生率或频率的明显剂量相关趋势。两名受试者停止了输注,一名由于轻度输注反应(第1部分,300mg组),且一名由于中度输注反应(第2部分,75mg组)。
生命指征、ecg或临床化学参数(包括中性粒细胞计数、血小板计数或c-反应蛋白水平)没有明显的剂量相关趋势或治疗相关的变化。
300mg组中的一名健康受试者,在给予后5-6周的随访中显示出非中和治疗急性抗肽1抗体。
总体而言,当以单次静脉注射剂量静脉给予高达750mg肽1、以及以单次皮下注射剂量以60mg给予肽1时,肽1是安全的且耐受性良好。
实施例3
临床试验000115(多个上升剂量)
设计
这是一个安慰剂对照、双盲、剂量组内随机的平行组试验,其目的在于研究多个上升剂量的肽1的安全性、耐受性和药代动力学。所研究的剂量为:通过静脉输注历经30分钟(75mg)或历经1小时(300mg和600mg),一周一次给予75、300和600mg肽1,持续4周。
包括二十四(24)名健康受试者,其中18名(11名男性和7名女性)接收活性剂治疗并且6名(2名男性和4名女性)接收安慰剂。
结果
pk评价显示,在第一个和最后一个治疗日的特性非常接近,并且类似于单剂量研究的结果。auc和c最大在第一次和第四次剂量给予后是剂量成比例的,在第一次剂量后的c最大浓度为19、78和148μg/ml,并且在第四次剂量后为19、79和142μg/ml(对于健康受试者中的单剂量,其为16、77和161μg/ml;图5)。对于三个剂量水平,相应的谷值分别为0.66、2.68、4.56μg/ml以及0.98、3.95和7.67μg/ml。最后剂量后计算的平均末期半衰期约为5.5天。
肽1的安全性良好,在包括安慰剂组在内的所有治疗组中发生的不良事件很少,且均为轻度或中度。没有观察到不良事件的发生率或频率的明显剂量相关趋势。一名受试者由于轻度输注反应而退出(600mg组)。
生命指征、ecg或临床化学参数(包括中性粒细胞计数、血小板计数或c-反应蛋白水平)没有明显的剂量相关趋势或治疗相关的变化。
在任何受试者中均未检测到抗肽1抗体。
总体而言,在给予4周,一周一次静脉注射给予高达600mg时,肽1是安全的且耐受性良好。
实施例4
药代动力学数据建模
使用2室结构模型可以充分描述来自000115试验的pk数据。图6中描绘了75、300和600mg肽1的预期曲线以及观察到的数据,并且表1中列出了估计的平均pk参数。
表1使用2室结构药代动力学模型的肽1的模型估计
在人体中,肽1与il-6/sil-6r复合物的结合亲和力为130pm。在75-600mg的剂量下,使用结合亲和力(kd;130pm)和il-6/sil-6r水平(c靶;基于sil-6r为2.0nm),在肽1的估计稳态水平下,占用水平是超过90%。
实施例5
肽1的制备
肽1在cho/dhfr-细胞中的克隆和表达
cho/dhfr-细胞获自欧洲细胞培养物保藏中心(ecacc,编号9406067)。贴壁cho/dhfr-细胞缺乏二氢叶酸还原酶(dhfr),其是一种催化叶酸还原为二氢叶酸,然后还原为四氢叶酸的酶。因此cho/dhfr-细胞显示出对抗叶酸药物氨甲蝶呤(mtx)的敏感性。
cho/dhfr-细胞系被充分表征和测试。对于微生物无菌、支原体和外来病毒,ecacc(portondown,uk)根据21cfr证实了cho/dhfr-亲本细胞系作为用于生产人类生物药物的细胞底物的安全性。
cdna序列的选择和构建
使用用于gp130的细胞外结构域(il6st,ncbi基因id3572,转录物变体1(np_002175),氨基酸23-617)和人igg1的fc结构域(ighg1,ncbi基因id3500,根据kabateu计数的氨基酸221-447)的序列,由geneartag(雷根斯堡,德国)合成了作为单个dna片段的肽1的单体(seqidno:1的多肽序列)的cdna序列。该序列经优化为最佳密码子以用于cho细胞中。将三个充分表征的点突变引入fc部分的下铰链区。
通过用cho细胞表达系统中已知功效的小鼠igg重链信号肽代替原始gp130信号肽,进一步修饰cdna序列。在蛋白质合成过程中切除信号肽。fc区中igg1cys-pro-pro-cys序列的存在导致两个相同的gp130-fc亚单元通过fc区上的巯基残基而二聚化,它们一起形成肽1。
图7显示了用于形成肽1的gp130-fc亚单元的核苷酸和氨基酸序列。
用于选择主细胞库(mcb)的表达质粒的构建
如下,将单体cdna克隆到pantvhg1表达载体(antitope)中,该表达载体含有用于mtx转染子选择的dhfr基因:首先,用mlui和eagi限制酶消化表达载体以允许插入肽1cdna。其次,使用ol1425和ol1426引物(表2)pcr扩增单体编码区并且使用mlui和eagi限制酶消化。再次,将消化的片段凝胶纯化并连接在一起以产生pfer02表达载体。在巨细胞病毒(cmv)启动子的控制下插入单体cdna。
表3示出了pfer02表达元素的功能。图8示出了pfer02表示元素的核苷酸序列。
表2用于扩增用于克隆到pantvhg1中的单体编码区的寡核苷酸序列
*单体1-特异性序列显示为大写字母,载体-特异性序列显示为小写字母,限制性位点加下划线
表3pfer02表达元素
导致最终肽1生产克隆的细胞系选择过程
将pfer02载体用平端限制酶sspi进行线性化,该pfer02载体具有位于β-内酰胺酶基因中的单个识别位点。使用脂质介导的转染,将直线化的质粒转移到5x106个cho/dhfr-细胞中。转染24小时后,在补充有5%透析胎牛血清(fcs)和100nm氨甲喋呤(mtx)的培养基中对转染的细胞进行选择。将转染的细胞以各种密度稀释到该培养基中并分配到96孔平底组织培养板中。在5%co2和37℃下在潮湿的气氛中培育细胞。在培育期间,定期添加新鲜的mtx选择培养基,以确保mtx水平和营养水平保持不变。
使用mtx选择的初始细胞系选择
转染后数周,使用genetix
生产力(pg/细胞/天)=((th–ti)/((vh+vi)/2))/时间
其中:
th是收获滴度[μg/ml]
ti是初始滴度[μg/ml]
vh是收获时的活细胞计数[x106个细胞/ml]
vi是最初的活细胞计数[x106个细胞/ml]
时间是ti和th之间经过的时间(天)。
基于生产力结果(pg/细胞/天),选择13个细胞系进行基因扩增。
用于肽1细胞系选择的mtx驱动的基因扩增
在增加浓度的mtx(0.1-50m)下通过选择性压力,对13个选择的细胞系进行第一轮基因扩增的选择。7-10天后,从13个细胞系的每一个的每一孔采样上清液并且检测肽1的滴度(elisa)。评估具有高肽1表达水平的每个细胞系的孔的生产力(pg/细胞/天)。使用总共16个来自显示生产力显著增加的细胞系的孔,开始进行第二轮基因扩增。
在存在增加的mtx浓度的情况下进行第二轮基因扩增;检测来自每个培养物的上清液的肽1滴度(elisa)。扩增来自每个细胞系的选定孔,并评估生产力(pg/细胞/天);确定了响应于增加的mtx选择压力而提高了生产力的五个细胞系。使用增加的mtx浓度下的选择压力,将这五个细胞系进行第三轮基因扩增;检测来自每个孔的上清液的肽1滴度(elisa)。对每个细胞系的选定孔进行扩增并且评估生产力(pg/细胞/孔);选择表现出高肽1表达的5个细胞系。
克隆的限制性稀释
在展示肽1表达的五个细胞系上进行限制性稀释克隆。在培育一周之后,使用genetix
细胞系对化学定义的培养基中的悬浮培养的适应
如下使16个细胞系适于在化学定义的培养基中的悬浮培养:首先,使贴壁培养中的选定细胞系适于悬浮在cho悬浮生长培养基(dmem高葡萄糖,包括l-谷氨酰胺和丙酮酸钠,5%透析的fcs,20mg/ll-脯氨酸,1x青霉素/链霉素,1%普兰尼克f68)中以及然后悬浮在化学定义的悬浮生长培养基(来自生命技术公司(lifetechnologiesltd.)(paisley,uk)的cd
一旦适应悬浮培养,分阶段使细胞系断奶到无血清的化学定义的悬浮生长培养基(cd
在无血清适应后,将16个细胞系进一步细化到5个克隆。评估这5个克隆的生长(细胞密度和细胞倍增时间)和生产力(pg/细胞/天),之后选择3个克隆。选择一个克隆以制备主细胞库。
进行主细胞库(mcb)和工作细胞库(wcb)的制备。将来自预种子原种的一个小瓶用于制备200小瓶mcb,并且使用一瓶mcb来制备200小瓶wcb。在每种情况下,将小瓶解冻,并通过离心除去冷冻保存培养基。将细胞重新悬浮并在生长培养基中体积繁殖(cd
当获得足够的细胞时,将细胞等分在冷冻保存培养基(92.5%cd
药物物质(ds)制造过程的描述
肽1ds制造过程的简要描述如下。在接种到生产生物反应器中之前,使用无蛋白质培养基,从wcb小瓶中复苏细胞并逐渐扩大。细胞培养完成后,通过培养物过滤除去细胞和细胞碎片。
纯化由三个色谱柱步骤(mabselectsure,sp琼脂糖,captoq或sartobind苯基)、一个浓缩和渗滤步骤组成并且包括两个特定的病毒减少/灭活步骤;tritonx-100(包膜病毒灭活)处理和纳滤步骤(去除包膜和非包膜病毒)。
浓缩和渗滤后,加入赋形剂以配制ds。将配制的肽1通过0.22μm过滤到容器中。
药品(dp)的描述和组成
dp是待通过静脉注射输注给予的无菌溶液。dp由处于ph7.6的含有25mml-组氨酸、200mm蔗糖和0.1mg聚山梨醇酯20/ml的等渗溶液中浓度为15mg/ml浓度的肽1组成。将小瓶用氮气覆盖以防止氧化。该产品旨在用于单次使用并且储存在-20℃下直至解冻用于临床给予。
组成和批次配方
表4中示出了药品的批次配方。
表4dp批次组成
*当前版本。
序列表
seqidno:1
seqidno:2
- 还没有人留言评论。精彩留言会获得点赞!