环状膦酸钠盐的晶体及其制造方法与流程
本发明涉及环状膦酸钠盐的晶体及其制造方法。
背景技术:
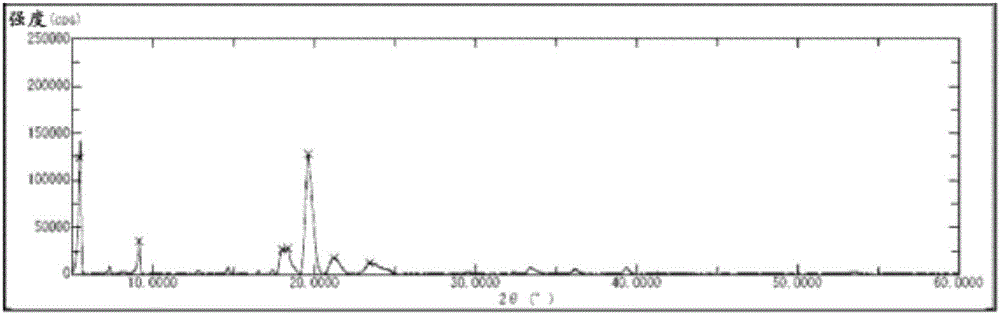
:下式(1)表示的环状膦酸钠盐(9-十八碳烯酸(9Z)-(2-羟基-2-氧代-2λ5-1,2-氧磷杂环戊烷-4-基)甲酯的钠盐)为一般被称为2ccPA的化合物。[化学式1]已知该2ccPA具有强效的镇痛作用(专利文献1),此外,还期待其被用作基于癌细胞的浸润抑制作用的抗癌剂(专利文献2)、基于透明质酸(hyaluronicacid)产生促进作用的变形性关节炎治疗药(专利文献3)等。一直以来,2ccPA是利用下述反应式-1所示的制造方法来制造的(专利文献2、专利文献4、非专利文献1及非专利文献2)。[化学式2][反应式-1]具体而言,首先,使利用非专利文献2中记载的制造方法而得到的碘化合物(5a)与亚磷酸三甲酯反应,得到膦酸二甲酯(6a)。接下来,使该化合物(6a)与对甲苯磺酸(p-TsOH)作用,得到化合物(8a)。向该化合物(8a)中导入油酸,制成化合物(9a),然后进行脱甲基化,进而制成钠盐,由此制得2ccPA。然而,上述制造方法中,由于未能实现各工序的反应条件的最优化、以及必须在各工序中实施利用硅胶柱色谱法的纯化,因此,由文献记载的收率计算,经过上述5个工序的2ccPA的总收率为18.7%,存在收率低、不适合大量合成的问题。此外,由于在脱甲基化工序中使用三甲基溴硅烷(TMSBr),因此,副反应生成的氢溴酸将导致反应体系中成为强酸性,有生成物分解的可能性。实际上,上述脱甲基化工序为38%的低收率。此外,最终工序中,利用氢氧化钠水溶液将化合物(10a)制成钠盐从而衍生成2ccPA,但由于没有特别地实施纯化就进行冷冻干燥,因此有在2ccPA固体中混入强碱性的氢氧化钠的可能性,无法避免由氢氧化钠导致的2ccPA的分解,在保存稳定性方面存在问题。因此,期望开发出与以往已知的方法相比更简便、并且能高收率地制造纯度高且保存稳定性优异的2ccPA晶体的方法。现有技术文献专利文献专利文献1:日本专利第5077893号公报专利文献2:日本特开2004-10582号公报专利文献3:国际公开第2013/069404号专利文献4:国际公开第03/104246号非专利文献非专利文献1:生物化学与生物物理学学报(BiochimicaetBiophysicaActa),2007,1771,p.103-112非专利文献2:Tetrahedoron,1991,Vol.47,No.6,p.1001-1012技术实现要素:发明要解决的课题本发明的课题在于提供纯度高且保存稳定性优异的2ccPA的晶体。此外,本发明的课题还在于提供简便且高收率地制造该2ccPA的晶体的方法。用于解决课题的手段本申请的发明人为了解决上述课题,反复进行了深入研究。结果,实现了通过1次硅胶柱色谱法的纯化而高收率地制造作为2ccPA的前体的环状膦酸酯,进而,成功地在不使用强酸或强碱的情况下由该环状膦酸酯衍生成2ccPA。此外,本申请的发明人发现以上述方式得到的2ccPA的晶体的保存稳定性优异,能够解决本课题。本发明是基于这样的见解完成的。即,本发明提供以下记载的2ccPA的晶体及其制造方法。项1.式(1)表示的环状膦酸钠盐(2ccPA)的晶体。[化学式3]项2.如项1所述的晶体,其在X射线粉末衍射光谱中,在由2θ表示的衍射角为15°~17°的位置具有特征峰。项3.如项1或2所述的晶体,其在X射线粉末衍射光谱中,在由2θ表示的衍射角为9°~10°的位置具有特征峰。项4.如项1~3中任一项所述的晶体,其在X射线粉末衍射光谱中,在由2θ表示的衍射角为3°~5°的位置具有特征峰。项5.如项4所述的晶体,其在X射线粉末衍射光谱中,在由2θ表示的衍射角为4.7°~5.0°的位置具有特征峰。项6.如项4所述的晶体,其在X射线粉末衍射光谱中,在由2θ表示的衍射角为4.4°~4.6°的位置具有特征峰。项7.如项4所述的晶体,其在X射线粉末衍射光谱中,在由2θ表示的衍射角为4.1°~4.3°的位置具有特征峰。项8.如项4所述的晶体,其在X射线粉末衍射光谱中,在由2θ表示的衍射角为3.7°~3.9°的位置具有特征峰。项9.如项1~8中任一项所述的晶体,其熔点为187~190℃。项10.项1~9中任一项所述的晶体的制造方法,所述制造方法包括下述工序,工序(H):使通式(9)表示的环状膦酸酯与卤化钠在有机溶剂中反应,从而得到2ccPA的工序;及工序(I):将含有工序(H)中得到的2ccPA的溶液在减压下浓缩、或将含有工序(H)中得到的2ccPA的溶液冷却,由此使晶体析出的工序。[化学式4](式中,R1表示烷基、芳基烷基或芳基。)项11.如项10所述的晶体的制造方法,所述制造方法还包括下述工序,工序(J):将工序(I)中得到的晶体溶解于水及/或有机溶剂中而得到溶液的工序;及工序(K):向工序(J)中得到的溶液中加入不良溶剂,由此进行重结晶的工序。项12.环状膦酸钠盐(2ccPA)的晶体,其是利用项10或11所述的制造方法而得到的。项13.制造式(1)表示的环状膦酸钠盐(2ccPA)的晶体的方法,所述制造方法包括工序(H):使通式(9)表示的环状膦酸酯与卤化钠在有机溶剂中反应的工序。[化学式5][化学式6](式中,R1表示烷基、芳基烷基或芳基。)项14.通式(9)表示的环状膦酸酯的制造方法,所述制造方法包括工序(G):使通式(8)表示的化合物与油酸化合物反应的工序。[化学式7](式中,R1表示烷基、芳基烷基或芳基。)[化学式8](式中,R1与上述相同。)项15.通式(8)表示的化合物的制造方法,所述制造方法包括工序(F):使碱与通式(7)表示的化合物作用的工序。[化学式9](式中,R1表示烷基、芳基烷基或芳基。)[化学式10](式中,2个R1相同或不同,表示烷基、芳基烷基或芳基。)项16.通式(7)表示的化合物的制造方法,所述制造方法包括工序(E):使酸与通式(6)表示的化合物作用的工序。[化学式11](式中,2个R1相同或不同,表示烷基、芳基烷基或芳基。)[化学式12](式中,R1与上述相同。2个R2相同或不同,表示烷基、环烷基或芳基。)项17.通式(6)表示的化合物的制造方法,所述制造方法包括工序(D):使通式(5)表示的化合物与亚磷酸二酯反应的工序。[化学式13](式中,2个R1相同或不同,表示烷基、芳基烷基或芳基。2个R2相同或不同,表示烷基、环烷基或芳基。)[化学式14](式中,R2与上述相同。X表示卤素原子。)项18.通式(5)表示的卤素化合物的制造方法,所述制造方法包括工序(C):使通式(4)表示的化合物与碱金属卤化物及/或碱土金属卤化物在碱的存在下反应的工序。[化学式15](式中,2个R2相同或不同,表示烷基、环烷基或芳基。X表示卤素原子。)[化学式16](式中,R2与上述相同。R3表示可被取代的烷基或可被取代的芳基。)项19.通式(4)表示的化合物的制造方法,所述制造方法包括工序(B):使通式(3)表示的化合物与磺酰卤化物反应的工序。[化学式17](式中,2个R2相同或不同,表示烷基、环烷基或芳基。R3表示可被取代的烷基或可被取代的芳基。)[化学式18](式中,R2与上述相同。)项20.通式(5)表示的化合物的制造方法,所述制造方法包括工序(B’):使通式(3)表示的化合物与卤化剂反应的工序。[化学式19](式中,2个R2相同或不同,表示烷基、环烷基或芳基。X表示卤素原子。)[化学式20](式中,R2与上述相同。)项21.如项10或13所述的晶体的制造方法,所述晶体的制造方法还包括项14~20中任一项所述的工序。发明的效果本发明的2ccPA的晶体的保存稳定性优异,即使将该晶体长时间保存也几乎不分解。通过按照本发明的制造方法,能够利用简便的手段高收率地制造高纯度的2ccPA晶体。具体而言,本发明的制造方法包含新型的制造方法,特别地,本发明可以以不在各个工序中进行分离纯化(叠缩(telescoping)法)的方式制造作为2ccPA的前体的环状膦酸酯。此外,本发明的制造方法可以在不使用强酸或强碱的情况下简便地由环状膦酸酯提供减小了纯度低下的风险、且稳定性优异的2ccPA晶体。附图说明图1是实施例1中得到的2ccPA的晶体的X射线粉末衍射光谱。图2是实施例2中得到的2ccPA的晶体的X射线粉末衍射光谱。图3是实施例3中得到的2ccPA的晶体的X射线粉末衍射光谱。图4是实施例4中得到的2ccPA的晶体的X射线粉末衍射光谱。图5是实施例5中得到的2ccPA的晶体的X射线粉末衍射光谱。图6是实施例6中得到的2ccPA的晶体的X射线粉末衍射光谱。图7是实施例7中得到的2ccPA的晶体的X射线粉末衍射光谱。图8是实施例8中得到的2ccPA的晶体的X射线粉末衍射光谱。图9是实施例9中得到的2ccPA的晶体的X射线粉末衍射光谱。图10是实施例10中得到的2ccPA的晶体的X射线粉末衍射光谱。图11是实施例11中得到的2ccPA的晶体的X射线粉末衍射光谱。图12是实施例12中得到的2ccPA的晶体的X射线粉末衍射光谱。图13是实施例13中得到的2ccPA的晶体的X射线粉末衍射光谱。图14是实施例14中得到的2ccPA的晶体的X射线粉末衍射光谱。图15是实施例15中得到的2ccPA的晶体的X射线粉末衍射光谱。图16是实施例16中得到的2ccPA的晶体的X射线粉末衍射光谱。图17是实施例17中得到的2ccPA的晶体的X射线粉末衍射光谱。图18是实施例18中得到的2ccPA的晶体的X射线粉末衍射光谱。图19是实施例19中得到的2ccPA的晶体的X射线粉末衍射光谱。图20是实施例20中得到的2ccPA的晶体的X射线粉末衍射光谱。图21是实施例21中得到的2ccPA的晶体的X射线粉末衍射光谱。图22是实施例22中得到的2ccPA的晶体的X射线粉末衍射光谱。图23是实施例23中得到的2ccPA的晶体的X射线粉末衍射光谱。图24是实施例24中得到的2ccPA的晶体的X射线粉末衍射光谱。图25是实施例25中得到的2ccPA的晶体的X射线粉末衍射光谱。图26是实施例26中得到的2ccPA的晶体的X射线粉末衍射光谱。图27是表示稳定性试验的结果的图。具体实施方式以下,详细地说明本发明的2ccPA的新型的晶体及其制造方法。本说明书中,关于用语“包含”,其包括“包含”、“实质上仅包含”及“仅包含”的概念。1.环状膦酸钠盐(2ccPA)的晶体本发明的2ccPA的晶体为下式(1)表示的环状膦酸钠盐(9-十八碳烯酸(9Z)-(2-羟基-2-氧代-2λ5-1,2-氧磷杂环戊烷-4-基)甲酯的钠盐,IUPAC名:4-[(Z)-十八碳-9-烯酰基氧基甲基]-2-氧代-1,2-λ5-氧磷杂环戊烷-2-醇钠盐)的晶体。[化学式21]对于该2ccPA的晶体而言,例如,使用株式会社Rigaku制的RINT-2000UltimaIV(商品名)进行测定的情况下,利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱在下述的晶面间距(latticespacing)(d)的位置具有特征峰。对于该2ccPA的晶体而言,在晶体X射线粉末衍射光谱中,以由2θ表示的衍射角而言,包含:在约15°~17°的位置处的特征峰(以下,称为“峰A”)、在约9°~10°的位置处的特征峰(以下,称为“峰B”)、或在约3°~5°的位置处的至少1个特征峰(以下,称为“峰C~F”)。该峰C~F还包含下述峰C、D、E及/或F。在约4.7°~5.0°的位置处的特征峰(以下,称为“峰C”)在约4.4°~4.6°的位置处的特征峰(以下,称为“峰D”)在约4.1°~4.3°的位置处的特征峰(以下,称为“峰E”)在约3.7°~3.9°的位置处的特征峰(以下,称为“峰F”)该峰C包含在约4.7°~4.9°的位置及/或约4.9°~5.0°的位置处的特征峰。本发明中的2ccPA的晶体实质上具有鳞片状晶形层叠而成的形状。本发明中的2ccPA的晶体的熔点在187℃~190℃的范围内。该熔点是使用BUCHI公司制B-545型熔点测定装置测定的。本发明中的2ccPA的晶体的IR光谱是使用PerkinElmer公司的IR测定装置SpectrumOneB测定的。本发明中的2ccPA的晶体的纯度是使用采用了反相硅胶柱的高效液相色谱法(HPLC)测定的。一般地,该纯度为98%以上。本发明的2ccPA的晶体的保存稳定性优异。即使将该2ccPA的晶体于-20℃及35℃密闭保存3个月,其纯度也均几乎不降低,不易发生2ccPA的分解。2.上述2ccPA的晶体的制造方法本发明的2ccPA的晶体的制造方法的特征在于,包括下述工序(H)及工序(I)。所述制造方法的特征在于,包括:工序(H):使通式(9)表示的环状膦酸酯与卤化钠在有机溶剂中反应,从而得到2ccPA的工序;及工序(I):将含有工序(H)中得到的2ccPA的溶液在减压下浓缩、或将含有工序(H)中得到的2ccPA的溶液冷却,由此使晶体析出的工序。[化学式22](式中,R1与上述相同。)进而,本发明的2ccPA的晶体的制造方法除了包括上述工序(H)及工序(I)以外,还可包括下述工序(J)及工序(K)。工序(J):将工序(H)及(I)中得到的晶体溶解于水及/或有机溶剂中的工序;及工序(K):向工序(J)中得到的溶液中加入不良溶剂,由此进行重结晶的工序。2-1.工序(H)工序(H)为下述反应式-2所示的工序。[化学式23][反应式-2](式中,R1与上述相同。)具体而言,工序(H)是使通式(9)表示的环状膦酸酯与卤化钠在有机溶剂中反应从而得到式(1)表示的2ccPA的工序,该工序(H)中,得到含有2ccPA的溶液。工序(H)中使用的上述通式(9)表示的环状膦酸酯是经过后述的制造工序而制造的。通式(9)表示的环状膦酸酯中,作为R1表示的烷基,例如,可举出链状或支链状的碳原子数为1~10的烷基,具体而言,可举出甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、正己基、正庚基、正辛基、正壬基等。优选为碳原子数为1~6的烷基,更优选为碳原子数为1~4的烷基,特别优选为甲基、乙基及异丙基。上述烷基可以具有1~5个、优选为1~3个的卤素原子(例如,氟、氯、溴等)、碳原子数为1~6的烷氧基、硝基等取代基。作为R1表示的芳基烷基,例如,可举出碳原子数为7~16的芳基烷基(芳基部分的碳原子数为6~10,烷基部分的碳原子数为1~6),具体而言,可举出苄基;1-苯基乙基、2-苯基乙基;1-苯基丙基、2-苯基丙基、3-苯基丙基;1-苯基丁基、2-苯基丁基、3-苯基丁基、4-苯基丁基;萘甲基等。优选为碳原子数为7~11的芳基烷基,更优选为碳原子数为7~8的芳基烷基,特别优选为苄基。构成R1表示的上述芳基烷基的芳基可具有1~5个、优选为1~3个的下述取代基:例如,卤素原子(例如,氟、氯、溴等)、碳原子数为1~6的烷基、碳原子数为1~6的烷氧基、硝基等。作为R1表示的上述芳基,例如,可举出单环或二环以上的芳基,具体而言,可举出苯基、萘基、蒽基、菲基等。其中优选为可具有取代基的苯基。该芳基可具有1~5个、优选为1~3个的下述取代基:例如,卤素原子(例如,氟、氯、溴等)、碳原子数为1~6的烷基、碳原子数为1~6的烷氧基、硝基等。作为工序(H)中使用的卤化钠,可广泛使用已知的卤化钠,例如,可举出氟化钠、氯化钠、溴化钠、碘化钠等。其中,优选碘化钠。卤化钠可单独使用1种或混合使用2种以上。相对于1摩尔上述通式(9)表示的化合物而言,该卤化钠的使用量通常为1~5摩尔,优选为1~3摩尔,更优选为1~1.5摩尔。作为工序(H)中使用的有机溶剂,只要是不对该反应造成不良影响的溶剂则没有特别限定。作为使用的有机溶剂,例如,可举出酮系溶剂(支链或直链状酮及环状酮,例如,丙酮、甲基乙基酮、甲基丁基酮、甲基异丁基酮、DIBK(二异丁基酮)、环己酮等)、醇系溶剂(例如,甲醇、乙醇等)、醚系溶剂(乙醚、二异丙醚、四氢呋喃(THF)、1,4-二氧杂环己烷等)、芳香族烃系溶剂(例如,苯、甲苯、二甲苯等)、脂肪族或脂环式烃系溶剂(正戊烷、正己烷、环己烷、石油醚等)、酯系溶剂(乙酸乙酯等)、卤代烃系溶剂(二氯甲烷、氯仿、1,2-二氯乙烯等)等。有机溶剂可单独使用1种或组合使用2种以上。这些有机溶剂中,优选酮系溶剂,特别优选丙酮、甲基乙基酮及甲基异丁基酮。作为有机溶剂的使用量,可在宽泛的范围内进行适宜选择,例如,相对于1摩尔通式(9)表示的化合物而言,一般为2~20升,优选为2~5升。工序(H)可在氮、氩等非活性气体的气氛下进行。反应压力没有特别限制,可在常压下或加压下实施。反应温度通常为0~120℃,优选为50~120℃,更优选为70~120℃。反应时间通常为0.1~100小时,优选为0.5~50小时,更优选为1~24小时。反应结束后,可利用浓缩、析晶、过滤等通常的分离方法将过量的试剂(例如,卤化钠)、未反应的原料化合物等从得到的反应混合物中除去,并取出作为目标的通式(1)表示的2ccPA。2-2.工序(I)工序(I)是将含有工序(H)中得到的2ccPA的溶液在减压下浓缩、或将含有工序(H)中得到的2ccPA的溶液冷却,由此使晶体析出的工序。对于工序(I)中的减压而言,只要为析出晶体的压力则没有特别限制,通常低于大气压即可。工序(I)中的冷却温度只要为使晶体析出的温度则没有特别限制,所述温度通常低于工序(H)的反应后的溶液的温度,优选为0~30℃,更优选为10~25℃。对于冷却时间而言,没有特别限制,通常为0.1~100小时,优选为0.5~50小时,更优选为1~2小时。可将得到的晶体用于接下来的工序(J)。2-3.工序(J)工序(J)是将工序(H)及(I)中得到的晶体溶解于水及/或有机溶剂中从而得到溶液的工序。工序(J)中使用的水及/或有机溶剂为能够将工序(I)中得到的晶体溶解的水及/或有机溶剂即可。作为该有机溶剂,例如,可举出醇系溶剂,特别优选甲醇、乙醇、1-丙醇、异丙醇、或1-丁醇。作为水及/或有机溶剂的使用量,可在宽泛的范围内进行适宜选择,例如,相对于1摩尔2ccPA而言,一般为0.5~20升,优选为0.5~2升。使用水与有机溶剂的混合溶剂的情况下,其配合比率没有特别限制,水与有机溶剂的配合比率优选为1:99~99:1,更优选为30:70~70:30。溶解的温度没有特别限定,通常为0~100℃,优选为10~80℃,更优选为20~60℃。工序(J)的时间没有特别限定,通常为0.1~100小时,优选为0.5~50小时,更优选为1~2小时。2-4.工序(K)工序(K)是向工序(J)中得到的溶液中加入不良溶剂,由此进行重结晶的工序。作为工序(K)中使用的不良溶剂,只要是能够使晶体从工序(J)中得到的溶液中析出的溶剂即可。具体而言,作为该不良溶剂,为相对于工序(J)中使用的溶剂(良溶剂)而言的不良溶剂即可,例如,可举出酮系溶剂(例如,丙酮、甲基乙基酮、甲基异丁基酮等)、醚系溶剂(乙醚、二异丙醚、四氢呋喃(THF)、1,4-二氧杂环己烷等)、芳香族烃系溶剂(例如,苯、甲苯、二甲苯等)、脂肪族或脂环式烃系溶剂(正戊烷、正己烷、环己烷、石油醚等)、酯系溶剂(乙酸甲酯、乙酸乙酯、乙酸异丙酯、乙酸丁酯等)、卤代烃系溶剂(二氯甲烷、氯仿、1,2-二氯乙烯等)、碳原子数为3以上的醇系溶剂(例如,1-丙醇)等。此外,该工序(K)中使用的溶剂只要是相对于工序(J)中使用的溶剂(良溶剂)而言的不良溶剂即可,例如,工序(K)中使用的溶剂为甲醇的情况下,作为该不良溶剂,可使用碳原子数为3以上的醇系溶剂(例如,1-丙醇)。有机溶剂可单独使用1种或组合使用2种以上。这些有机溶剂中,优选酮系溶剂,特别优选丙酮、甲基乙基酮及甲基异丁基酮。作为不良溶剂的使用量,可从宽泛的范围内进行适宜选择,例如,相对于1摩尔2ccPA而言,一般为1~30升,优选为2~5升。加入不良溶剂时的温度通常为-20℃~30℃,优选为-10℃~20℃,更优选为0℃~20℃。利用包括上述工序(H)及工序(I)的制造方法而得到的、或利用包括上述工序(H)~工序(K)的制造方法而得到的环状膦酸钠盐(2ccPA)的晶体具有纯度高、且保存稳定性优异的优点。3.通式(9)表示的环状膦酸酯的制造方法本发明中的通式(9)表示的环状膦酸酯可经过下述反应式-3所示的工序来制造。[化学式24][反应式-3](式中,R1、R2、R3、及X与上述相同。)以下,针对上述工序(A)至工序(G)详细地进行说明。3-1.工序(A):(缩醛保护工序)工序(A)为下述反应式-4所示的工序。[化学式25][反应式-4](式中,R2与上述相同。)具体而言,工序(A)为使上式(2)表示的2-羟甲基-1,3-丙二醇与酮化合物或缩醛化合物在酸的存在下反应,从而制造通式(3)表示的环状缩醛化合物的工序(缩醛保护工序)。作为工序(A)中使用的酮化合物,只要是具有酮基的有机化合物,则没有特别限制。作为该酮化合物,例如,可举出下述通式(10)表示的酮化合物。[化学式26](式中,R2与上述相同。2个R2可彼此键合而形成亚烷基,该亚烷基可以进一步具有取代基。)作为工序(A)中使用的缩醛化合物,没有特别限制,可举出例如下述通式(11)表示的缩醛化合物。[化学式27](式中,R2与上述相同。2个R2可彼此键合而形成亚烷基,该亚烷基可以进一步具有取代基。2个R4相同或不同,表示烷基。)通式(10)表示的酮化合物或通式(11)表示的缩醛化合物中,作为R2表示的烷基,例如,可举出链状或支链状的碳原子数为1~10的烷基,具体而言,可举出甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、正己基、正庚基、正辛基、正壬基等。优选碳原子数为1~6的烷基,更优选碳原子数为1~4的烷基,特别优选为甲基、乙基及异丙基。该烷基可具有1~5个、优选为1~3个的下述取代基:例如,卤素原子(例如,氟、氯、溴等)、芳基(例如,苯基、萘基等)、羧基等。通式(10)表示的酮化合物或通式(11)表示的缩醛化合物中,作为R2表示的可具有取代基的环烷基中的环烷基,例如,可举出碳原子数为3~10的环烷基,具体而言,可举出环丙基、环丁基、环戊基、环己基、环庚基、环辛基等。优选碳原子数为3~7的环烷基,更优选碳原子数为5~7的环烷基,特别优选为环己基。该环烷基可具有1~5个、优选为1~3个的下述取代基:例如,卤素原子(例如,氟、、氯、溴等)、烷基(碳原子数为1~6的烷基)、芳基(例如,苯基、萘基等)、羧基等。通式(10)表示的酮化合物或通式(11)表示的缩醛化合物中,作为R2表示的可具有取代基的芳基中的芳基,例如,可举出单环或二环以上的芳基,具体而言,可举出苯基、萘基、蒽基、菲基等。其中,优选为可具有取代基的苯基。该芳基可具有1~5个、优选为1~3个的下述取代基:例如,卤素原子(例如,氟、氯、溴等)、烷基(碳原子数为1~6的烷基)、羧基等。上述通式(10)或(11)中,2个R2可彼此键合而形成亚烷基,该亚烷基可具有取代基。2个R2彼此键合而形成亚烷基的情况下,作为该亚烷基,例如,可举出-(CH2)q-(q为1~6的整数)、-(CH=CH)r-(r表示1、2或3)、-CH=CH-(CH2)s-(s表示1、2或3)等。该亚烷基可具有取代基,作为该取代基,例如,可举出烷基(例如,碳原子数为1~6的烷基)、芳基(例如,苯基、萘基)、氧代基(=O)、卤素原子(例如,氟原子、氯原子、溴原子、碘原子)等。在二价烃基上可具有选自由它们组成的组中的1~5个取代基。通式(11)表示的缩醛化合物中,作为R4表示的烷基,例如,可举出链状或支链状的碳原子数为1~10的烷基,具体而言,可举出甲基、乙基,正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、正己基、正庚基、正辛基、正壬基等。优选碳原子数为1~6的烷基,更优选碳原子数为1~4的烷基,特别优选为甲基、乙基及异丙基。该烷基可具有1~5个、优选为1~3个的下述取代基:例如,卤素原子(例如,氟、氯、溴等)、芳基(例如,苯基、萘基等)、羧基等。作为工序(A)中使用的酮化合物,具体而言,例如,可举出丙酮、2-丁酮(甲基乙基酮)、2-戊酮、3-戊酮、4-甲基-2-戊酮、甲基异丙基酮、甲基异丁基酮、2-己酮、3-己酮、2-庚酮、3-庚酮、2-辛酮、3-辛酮、2-壬酮、2-癸酮、4-癸酮、2-十一酮、6-十一酮等碳原子数为3~20的链状脂肪族酮化合物;2-甲基环己酮、3-甲基环己酮、3-甲基环戊酮、4-乙酰基-1-甲基环己烯等碳原子数为6~20的环状脂肪族酮化合物;苯乙酮、1-(4-氯苯基)-1-乙酮、1-(2-氯苯基)-1-乙酮、1-(4-氟苯基)-1-乙酮、1-(2-氟苯基)-1-乙酮、1-(4-甲基苯基)-1-乙酮、1-(2-甲基苯基)-1-乙酮、1-(4-硝基苯基)-1-乙酮、1-(4-叔丁基苯基)-1-乙酮、1-(4-甲氧基苯基)-1-乙酮、1-(4-烯丙氧基羰基苯基)-1-乙酮、1-苯基-2-丙酮、4-氧代-4-苯基丁酸甲酯、4-氧代-4-苯基丁酸乙酯、1-苯基-2-丁酮、4-苯基-2-丁酮、2-苯基环戊酮、2-苯基环庚酮、9-乙酰基蒽、2-乙酰基联苯、4-乙酰基联苯、2-乙酰基萘、2-乙酰基菲、3-乙酰基菲、9-乙酰基菲等碳原子数为6~20的芳香族酮化合物;2-乙酰基-5-降冰片烯等芳烷基酮化合物等。这些化合物中,可举出丙酮、2-戊酮、3-戊酮、甲基乙基酮、甲基异丙基酮、甲基异丁基酮、环丁酮、环戊酮、环己酮等。特别优选丙酮、甲基乙基酮、及甲基异丁基酮。作为本发明中使用的缩醛化合物,具体而言,例如,可举出2,2-二甲氧基丙烷、2,2-二乙氧基丙烷、2,2-二丙氧基丙烷、2,2-二丁氧基丙烷、1,1-二甲氧基环己烷、1,1-二甲氧基环戊烷、苯甲酮二甲基缩酮、2,2-二甲基-1,3-二氧戊环、4,4-二甲氧基庚烷、5,5-二甲氧基壬烷、4,4-二乙氧基庚烷、5,5-二乙氧基壬烷等。特别优选2,2-二甲氧基丙烷、2,2-二丙氧基丙烷、2,2-二丁氧基丙烷、及苯甲酮二甲基缩酮。作为工序(A)中使用的酸,可举出已知的无机酸及有机酸。作为无机酸,可举出盐酸、硫酸等。作为有机酸,可举出甲磺酸、三氟甲磺酸、苯磺酸、对甲苯磺酸、吡啶对甲苯磺酸盐等磺酸化合物;乙酸等羧酸化合物等。作为酸,特别优选盐酸、硫酸、对甲苯磺酸、吡啶对甲苯磺酸盐、乙酸等。这里,使用乙酸时,可兼用作溶剂。作为酸的使用量,可从宽泛的范围内进行适宜选择,例如,相对于1摩尔上式(2)表示的2-羟甲基-1,3-丙二醇而言,一般为0.01~500摩尔,优选为0.01~2摩尔,更优选为0.01~1摩尔。工序(A)是在无溶剂或溶剂的存在下实施的。使用溶剂的情况下,作为该溶剂,只要不对该反应造成不良影响则没有特别限定。作为使用的溶剂,例如,可举出醇系溶剂(例如,甲醇、乙醇等)、醚系溶剂(乙醚、二异丙醚、四氢呋喃(THF)、1,4-二氧杂环己烷等)、芳香族烃系溶剂(例如,苯、甲苯、二甲苯等)、脂肪族或脂环式烃系溶剂(正戊烷、正己烷、环己烷、石油醚等)、酯系溶剂(乙酸乙酯等)、卤代烃系溶剂(二氯甲烷、氯仿、1,2-二氯乙烯等)等。溶剂可单独使用1种或组合使用2种以上。这些溶剂中,优选甲醇、THF、1,4-二氧杂环己烷及甲苯,特别优选THF。作为溶剂的使用量,可从宽泛的范围内进行适宜选择,例如,相对于1摩尔通式(2)表示的化合物而言,一般为0~20升,优选为0~5升。工序(A)可在氮、氩等非活性气体的气氛下进行。反应压力没有特别限制,可在常压下或加压下实施。反应温度通常为0~100℃,优选为10~80℃,更优选为20~80℃。反应时间通常为0.1~100小时,优选为0.5~50小时,更优选为1~10小时。反应结束后,可利用分液、蒸馏、柱纯化等通常的分离方法将过量的试剂(酮化合物等)、未反应的原料化合物等从得到的反应混合物中除去,并取出作为目标的通式(3)表示的环状缩醛化合物。此外,反应结束后,也可仅进行浓缩、而不经过纯化及分离工序地将反应后的混合物直接用于工序(B)(叠缩合成)。3-2.工序(B):(磺酰化工序)工序(B)为下述反应式-5所示的工序。[化学式28][反应式-5](式中,R2及R3与上述相同。)工序(B)为使上述通式(3)表示的化合物与磺酰卤化物反应,从而得到通式(4)表示的化合物的工序(磺酰化工序)。作为工序(B),例如,可举出使上述通式(3)表示的醇化合物与磺酰卤化物在有机溶剂中、在碱的存在下反应,由此得到通式(4)表示的磺酸酯化合物的工序。使用例如甲磺酰氯作为磺酰化合物的反应可参考非专利文献2中记载的方法。作为工序(B)中使用的磺酰卤化物,可举出甲磺酰氯、甲磺酰溴、甲磺酰碘等烷基磺酰卤化物;苯磺酰氯、对甲苯磺酰氯等芳基磺酰卤化物等。作为磺酰卤化物的使用量,可从宽泛的范围内进行适宜选择,例如,相对于1摩尔上式(3)表示的甲醇化合物而言,一般为1~500摩尔,优选为1~10摩尔,更优选为1~2摩尔。工序(B)是在无溶剂或溶剂的存在下实施的。使用溶剂的情况下,作为该溶剂,只要不对该反应造成不良影响则没有特别限定。作为使用的溶剂,例如,可举出醇系溶剂(例如,甲醇、乙醇等)、醚系溶剂(乙醚、二异丙醚、四氢呋喃(THF)、1,4-二氧杂环己烷等)、芳香族烃系溶剂(例如,苯、甲苯、二甲苯等)、脂肪族或脂环式烃系溶剂(正戊烷、正己烷、环己烷、石油醚等)、酯系溶剂(乙酸乙酯等)、卤代烃系溶剂(二氯甲烷(MDC,DCM)、氯仿、1,2-二氯乙烯等)等。溶剂可单独使用1种或组合使用2种以上。这些溶剂中,优选THF、1,4-二氧杂环己烷、甲苯及二氯甲烷,特别优选二氯甲烷。作为溶剂的使用量,可从宽泛的范围内进行适宜选择,例如,相对于1摩尔通式(2)表示的化合物而言,一般为0~20升,优选为1~5升。工序(B)可在氮、氩等非活性气体的气氛下进行。反应压力没有特别限制,可在常压下或加压下实施。反应温度通常为-40~100℃,优选为-30~80℃,更优选为-20~20℃。反应时间通常为0.1~100小时,优选为0.5~50小时,更优选为1~4小时。反应结束后,可利用分液、浓缩、柱纯化等通常的分离方法将过量的试剂(磺酰卤化物等)、未反应的原料化合物等从得到的反应混合物中除去,并取出作为目标的通式(4)表示的环状缩醛化合物。此外,反应结束后,也可仅进行分液及浓缩、而不经过纯化及分离工序地将反应后的混合物直接用于工序(C)(叠缩合成)。3-3.工序(C):(卤化工序)工序(C)为下述反应式-6所示的工序。[化学式29][反应式-6](式中,R2、R3及X与上述相同。)具体而言,工序(C)为使上述通式(4)表示的化合物与碱金属卤化物及/或碱土金属卤化物在碱的存在下反应,从而得到通式(5)表示的化合物的工序(卤化工序)。作为工序(C)中使用的碱金属卤化物,没有特别限制,例如,可举出卤化锂(例如,氟化锂、氯化锂、溴化锂、碘化锂等)、卤化钠(例如,氟化钠、氯化钠、溴化钠、碘化钠等)、卤化钾(例如,氟化钾、氯化钾、溴化钾、碘化钾等)、卤化铯(例如,氟化铯、氯化铯、溴化铯、碘化铯等)等。其中,优选碘化钠。碱金属卤化物可单独使用1种或组合使用2种以上。作为工序(C)中使用的碱土金属卤化物,没有特别限制,例如,可举出卤化镁(例如,氟化镁、氯化镁、溴化镁、碘化镁等)、卤化钙(例如,氟化钙、氯化钙、溴化钙、碘化钙等)、卤化锶(例如,氟化锶、氯化锶、溴化锶、碘化锶等)、卤化钡(例如,氟化钡、氯化钡、溴化钡、碘化钡等)等。碱土金属卤化物可单独使用1种或组合使用2种以上。作为碱金属卤化物及/或碱土金属卤化物的使用量,相对于1摩尔通式(4)表示的化合物而言,通常为1摩尔以上,优选为1~10摩尔,更优选为1~3摩尔。作为工序(C)中使用的碱,例如,可举出有机碱及无机碱。作为有机碱,例如,可举出三甲胺、三乙胺、三丁胺、二异丙基乙胺等具有1~3个(优选为3个)的碳原子数为1~4的烷基的有机胺。特别优选三乙胺。作为无机碱,具体而言,可举出碳酸氢钠、碳酸钠、碳酸氢钾、碳酸钾、碳酸钙等碱金属及碱土金属的碳酸盐。特别优选为碳酸氢钠。作为碱的使用量,可为催化量,例如,相对于上述通式(4)表示的磺酸酯化合物而言,通常为0.01摩尔以上,优选为0.01~1摩尔,更优选为0.05~0.5摩尔。工序(C)中,通过加入催化量的该碱,可防止分解反应,能够以高收率得到通式(5)表示的卤素化合物。工序(C)是在无溶剂或溶剂的存在下实施的。使用溶剂的情况下,作为该溶剂,只要不对该反应造成不良影响则没有特别限定。作为使用的溶剂,例如,可举出酮系溶剂(例如,丙酮、甲基乙基酮、甲基异丁基酮等)、醇系溶剂(例如,甲醇、乙醇等)、醚系溶剂(乙醚、二异丙醚、四氢呋喃(THF)、1,4-二氧杂环己烷等)、芳香族烃系溶剂(例如,苯、甲苯、二甲苯等)、脂肪族或脂环式烃系溶剂(正戊烷、正己烷、环己烷、石油醚等)、酯系溶剂(乙酸乙酯等)、卤代烃系溶剂(二氯甲烷、氯仿、1,2-二氯乙烯等)等。溶剂可单独使用1种或组合使用2种以上。这些溶剂中,特别优选丙酮、甲基乙基酮及甲基异丁基酮。作为溶剂的使用量,可从宽泛的范围内进行适宜选择,例如,相对于1摩尔通式(4)表示的化合物而言,一般为0~20升,优选为1~5升。工序(C)可在氮、氩等非活性气体的气氛下进行。反应压力没有特别限制,可在常压下或加压下实施。反应温度通常为0~120℃,优选为10~100℃,更优选为55~80℃。反应时间通常为0.1~100小时,优选为0.5~50小时,更优选为1~18小时。本反应为在卤化反应中使用碱的新型的反应。反应结束后,可利用分液、浓缩、柱纯化等通常的分离方法将过量的试剂(碱金属卤化物、碱土金属类、碱等)、未反应的原料化合物等从得到的反应混合物中除去,并取出作为目标的通式(5)表示的化合物。此外,反应结束后,也可不经过纯化及分离工序地将反应的混合物直接用于工序(D)(叠缩合成)。3-4.工序(B’):(其他方法的卤化工序)工序(B’)为下述反应式-7所示的工序。[化学式30][反应式-7](式中,R2及X与上述相同。)具体而言,工序(B’)为使上述通式(3)表示的环状缩醛化合物与卤化剂反应的工序。作为工序(B’)中使用的卤化剂,没有特别限制。例如,氯化的情况下,可举出氯、亚硫酰氯、三氯化磷、五氯化磷、三苯基膦-四氯化碳、三苯基膦-N-氯代丁二酰亚胺等。溴化的情况下,可举出溴、氢溴酸、三溴化磷、三苯基膦-溴、三苯基膦-N-溴代丁二酰亚胺、三苯基膦-四溴化碳、亚硫酰溴等。碘化的情况下,可举出碘、三苯基膦-碘、三苯基膦-N-碘代丁二酰亚胺等。其中,优选三苯基膦-碘或三苯基膦-四溴化碳。作为卤化剂的使用量,相对于1摩尔通式(3)表示的醇化合物而言,通常为1~500摩尔,优选为1~10摩尔,更优选为1~2摩尔。对于工序(B’)而言,为了捕捉反应中生成的酸,可在咪唑的存在下使其反应。作为咪唑的使用量,例如,相对于上述通式(3)表示的环状缩醛化合物而言,通常为1~500摩尔,优选为1~10摩尔,更优选为1~2摩尔。工序(B’)是在无溶剂或溶剂的存在下实施的。使用溶剂的情况下,作为该溶剂,只要不对该反应造成不良影响则没有特别限定。作为使用的溶剂,例如,可举出酮系溶剂(例如,丙酮、甲基乙基酮、甲基异丁基酮等)、醇系溶剂(例如,甲醇、乙醇等)、醚系溶剂(乙醚、二异丙醚、环戊基甲醚(CPME)、四氢呋喃(THF)、1,4-二氧杂环己烷等)、芳香族烃系溶剂(例如,苯、甲苯、二甲苯等)、脂肪族或脂环式烃系溶剂(正戊烷、正己烷、环己烷、石油醚等)、酯系溶剂(乙酸乙酯等)、卤代烃系溶剂(二氯甲烷、氯仿、1,2-二氯乙烯等)等。溶剂可单独使用1种或组合使用2种以上。这些溶剂中,特别优选丙酮、甲基乙基酮及甲基异丁基酮。作为溶剂的使用量,可从宽泛的范围内进行适宜选择,例如,相对于1摩尔通式(4)表示的化合物而言,一般为0~20升,优选为1~5升。工序(B’)可在氮、氩等非活性气体的气氛下进行。反应压力没有特别限制,可在常压下或加压下实施。反应温度通常为0~100℃,优选为0~40℃,更优选为0~20℃。反应时间通常为0.1~100小时,优选为0.5~50小时,更优选为1~5小时。反应结束后,可利用分液、浓缩、柱纯化等通常的分离方法将过量的试剂(卤化剂等)、未反应的原料化合物等从得到的反应混合物中除去,并取出作为目标的通式(5)表示的化合物。此外,反应结束后,也可不经过纯化及分离工序地将反应后的混合物直接用于工序(D)(叠缩合成)。3-5.工序(D):(膦酸二酯化工序)工序(D)为下述反应式-8所示的工序。[化学式31][反应式-8](式中,R1、R2及X与上述相同。)具体而言,工序(D)为使上述通式(5)表示的化合物与亚磷酸二酯在碱的存在下反应,从而得到通式(6)表示的化合物的工序(膦酸二酯化工序)。作为工序(D)中使用的亚磷酸二酯,可举出下述通式(12)表示的化合物。[化学式32](式中,R1与上述相同。)通式(12)表示的亚磷酸二酯中,作为R1表示的烷基、芳基烷基或芳基,与上述通式(9)表示的环状膦酸酯中举出的R1表示的烷基、芳基烷基或芳基相同。作为该亚磷酸二酯,具体而言,可举出例如亚磷酸二甲酯、亚磷酸二乙酯、亚磷酸二丙酯、亚磷酸二丁酯、亚磷酸二异丙酯、亚磷酸甲乙酯等亚磷酸二烷基酯;亚磷酸二苄酯、亚磷酸二苯乙酯等亚磷酸二芳基烷基酯;亚磷酸二苯酯、亚磷酸二甲苯酯等亚磷酸二芳基酯等,优选为亚磷酸二甲酯、亚磷酸二乙酯、亚磷酸二苄酯、及亚磷酸二苯酯。作为亚磷酸二酯的使用量,没有特别限制,例如,相对于上述通式(5)表示的卤素化合物而言,优选为1~10当量的范围,特别优选为2~2.5当量。作为工序(D)中使用的溶剂,只要为有机溶剂,则没有特别限制,例如,可使用非质子性极性溶剂。作为该非质子性极性溶剂,例如,可举出N,N-二甲基甲酰胺(DMF)、N,N-二甲基乙酰胺(DMAc)、N-甲基吡咯烷酮(NMP)、1,3-二甲基-2-咪唑啉酮等酰胺溶剂、二甲基亚砜(DMSO)、六甲基磷酰三胺(HMPA)、乙腈(AN)、丙酮、THF等。特别优选DMF、DMAc及乙腈。这些溶剂可单独使用1种或组合使用2种以上。作为溶剂的使用量,可从宽泛的范围内进行适宜选择,例如,相对于1摩尔通式(5)表示的化合物而言,一般为0~20升,优选为1~5升。作为工序(D)中使用的碱,例如,可举出有机碱及无机碱。作为有机碱,例如,可举出三甲胺、三乙胺、三丁胺、二异丙基乙胺等具有1~3个(优选为3个)的碳原子数为1~4的烷基的有机胺。特别优选为三乙胺。作为无机碱,具体而言,可举出碳酸氢钠、碳酸钠、碳酸氢钾、碳酸钾、碳酸铷、碳酸钙、碳酸铯等碱金属及碱土金属的碳酸盐。特别优选碳酸铯及碳酸铷。作为碱的使用量,没有特别限制,相对于上式(5)表示的化合物而言,优选为1~10当量的范围,特别优选为2~2.5当量。工序(D)可在氮、氩等非活性气体的气氛下进行。反应压力没有特别限制,可在常压下或加压下实施。反应温度通常为0~120℃,优选为20~80℃,更优选为40~50℃。反应时间通常为0.1~100小时,优选为0.5~50小时,更优选为5~8小时。反应结束后,可利用浓缩、过滤、柱纯化等通常的分离方法将过量的试剂(亚磷酸二酯、碱等)、未反应的原料化合物等从得到的反应混合物中除去,并取出作为目标的通式(6)表示的化合物。此外,反应结束后,也可仅进行浓缩及过滤、而不经过纯化及分离工序地将反应后的混合物直接用于工序(E)(叠缩合成)。3-6.工序(E):(开环工序)工序(E)为下述反应式-9所示的工序。[化学式33][反应式-9](式中,R1及R2与上述相同。)具体而言,工序(E)为使酸与通式(6)表示的化合物作用,从而得到通式(7)表示的化合物的工序(开环工序)。作为酸,可任意地使用已知的有机酸及无机酸。作为该有机酸,例如,可举出对甲苯磺酸(p-TsOH)、对甲苯磺酸吡啶鎓(PPTS)、樟脑磺酸(CSA)等磺酸、甲酸、乙酸、丙酸、丁酸、三氟乙酸(TFA)等碳原子数为1~4的低级脂肪酸等。特别优选p-TsOH及CSA。作为无机酸,具体而言,可举出盐酸、硫酸、硝酸等。特别优选盐酸。作为酸的使用量,没有特别限制,相对于1摩尔上式(6)表示的化合物而言,优选为0.01~0.2摩尔的范围,特别优选为0.01~0.1摩尔。工序(E)是在无溶剂或溶剂的存在下实施的。使用溶剂的情况下,作为该溶剂,只要不对该反应造成不良影响则没有特别限定。作为使用的溶剂,例如,可举出水、醇系溶剂(例如,甲醇、乙醇、正丙醇、异丙醇、正丁醇等碳原子数为1~4的低级醇)、醚系溶剂(乙醚、二异丙醚、四氢呋喃(THF)、1,4-二氧杂环己烷等)、芳香族烃系溶剂(例如,苯、甲苯、二甲苯等)、脂肪族或脂环式烃系溶剂(正戊烷、正己烷、环己烷、石油醚等)、酯系溶剂(乙酸乙酯等)、卤代烃系溶剂(二氯甲烷、氯仿、1,2-二氯乙烯等)等。溶剂可单独使用1种或组合使用2种以上。这些溶剂中,优选甲醇、THF、1,4-二氧杂环己烷及甲苯,特别优选甲醇。作为溶剂的使用量,可从宽泛的范围内进行适宜选择,例如,相对于1摩尔通式(6)表示的化合物而言,一般为0~20升,优选为1~5升。工序(E)可在氮、氩等非活性气体的气氛下进行。反应压力没有特别限制,可在常压下或加压下实施。反应温度通常为0~120℃,优选为0~80℃,更优选为0~20℃。反应时间通常为0.1~100小时,优选为0.5~50小时,更优选为3~12小时。反应结束后,可利用浓缩、柱纯化等通常的分离方法将过量的试剂(酸等)、未反应的原料化合物等从得到的反应混合物中除去,并取出作为目标的通式(7)表示的化合物。此外,反应结束后,也可仅进行浓缩、而不经过纯化及分离工序地将反应后的混合物直接用于工序(F)(叠缩合成)。3-7.工序(F):(环化工序)工序(F)为下述反应式-10所示的工序。[化学式34][反应式-10](式中,R1与上述相同。)具体而言,工序(F)为使碱与通式(7)表示的化合物作用,从而得到通式(8)的化合物的工序(环化工序)。作为工序(F)中使用的碱,可任意地使用已知的有机碱及无机碱。例如,作为有机碱,可举出1,8-二氮杂双环[5.4.0]-十一碳-7-烯(diazabicycloundecene,DBU)、1,5-二氮杂双环[4.3.0]-壬-5-烯(diazabicyclononene,DBN)、三甲胺、三乙胺(TEA)、三丁胺、二异丙基乙胺(DIPEA)等有机叔胺;甲醇钠(NaOMe)、乙醇钠(NaOEt)、叔丁醇钾(t-BuOK)、叔丁醇钠(t-BuONa)等金属醇盐等。作为无机碱,可举出碳酸铯(Cs2CO3)、碳酸铷(Rb2CO3)等碱金属碳酸盐;氢化钠(NaH)等碱金属氢化物等。作为碱,优选为DBU、DBN、TEA、DIPEA、NaOMe、NaOEt、t-BuOK、t-BuONa、Cs2CO3及NaH,更优选为DBU及DBN。作为碱的使用量,没有特别限制,例如,相对于1摩尔上述通式(7)表示的化合物而言,优选为0.1~2摩尔的范围,特别优选为0.1~1摩尔。工序(F)是在无溶剂或溶剂的存在下实施的。使用溶剂的情况下,作为该溶剂,只要不对该反应造成不良影响则没有特别限定。作为使用的溶剂,例如,可举出水、醇系溶剂(例如,甲醇、乙醇、正丙醇、异丙醇、正丁醇等碳原子数为1~4的低级醇);醚系溶剂(乙醚、二异丙醚(IPE)、四氢呋喃(THF)、1,4-二氧杂环己烷等);芳香族烃系溶剂(例如,苯、甲苯、二甲苯等);脂肪族或脂环式烃系溶剂(正戊烷、正己烷、环己烷、石油醚等);酯系溶剂(乙酸乙酯等);卤代烃系溶剂(二氯甲烷、氯仿、1,2-二氯乙烯等);酰胺系溶剂(例如,N,N-二甲基甲酰胺(DMF)、N,N-二甲基乙酰胺(DMAc)、N-甲基吡咯烷酮(NMP)、1,3-二甲基-2-咪唑啉酮等);二甲基亚砜(DMSO);六甲基磷酰三胺(HMPA);乙腈(AN);丙酮等,溶剂可单独使用1种或组合使用2种以上。这些溶剂中,优选DMF、DMAc、AN、丙酮、甲醇、IPE及丁醇,特别优选DMF及DMAc。作为溶剂的使用量,可从宽泛的范围内进行适宜选择,例如,相对于1摩尔通式(7)表示的化合物而言,一般为0~20升,优选为1~5升。此外,工序(F)的反应中,为了使反应停止,可使用淬灭剂(quenchingagent)。作为淬灭剂,可使用已知的淬灭剂,例如,可使用有机酸。作为该有机酸,例如,可举出p-TsOH、CSA等磺酸、甲酸、乙酸、丙酸、丁酸、三氟乙酸等碳原子数为1~4的脂肪酸。特别优选p-TsOH及CSA。作为该淬灭剂的使用量,优选为与反应中加入的有机碱相同的摩尔量。工序(F)可在氮、氩等非活性气体的气氛下进行。反应压力没有特别限制,可在常压下或加压下实施。反应温度通常为0~60℃,优选为0~40℃,更优选为15~25℃。反应时间通常为0.1~100小时,优选为0.5~50小时,更优选为2~7小时。反应结束后,可利用浓缩、柱纯化等通常的分离方法将过量的试剂(碱等)、未反应的原料化合物等从得到的反应混合物中除去,并取出作为目标的通式(8)表示的化合物。此外,反应结束后,也可仅进行浓缩、而不经过纯化及分离工序地将反应后的混合物直接用于工序(G)(叠缩合成)。3-8.工序(G)(酯化工序)工序(G)为下述反应式-11所示的工序。[化学式35][反应式-11](式中,R1与上述相同。)具体而言,工序(G)为使通式(8)表示的环状膦酸化合物与油酸化合物反应、从而得到通式(9)的化合物的工序(酯化工序),其可适宜地应用已知的酯化反应。作为油酸化合物,可举出例如油酸、以及油酰卤化物、油酸酐、油酸酯等油酸衍生物。这些油酸化合物可单独使用1种或混合使用2种以上。作为工序(G)中使用的油酰卤化物中的卤素,可举出氯原子、溴原子或碘原子等。特别地,作为该卤素,优选氯原子。作为工序(G)中使用的油酸酯,可举出甲酯、乙酯等。作为油酸化合物的使用量,没有特别限制,例如,相对于1摩尔上述通式(8)表示的化合物而言,优选为1~2摩尔的范围,特别优选为1~1.5摩尔。作为工序(G),例如,可举出:使环状膦酸化合物(8)与油酸在缩合剂的存在下反应的方法(工序G-1);使环状膦酸化合物(8)与油酰卤化物在碱的存在下反应的方法(工序G-2);使环状膦酸化合物(8)与油酸酐反应的方法(工序G-3);使环状膦酸化合物(8)与油酸酯反应的方法(工序G-4);等等。作为工序G-1中使用的缩合剂,只要为已知的缩合剂则没有限制,例如,可举出二环己基碳二亚胺(DCC)、二异丙基碳二亚胺(DIC)、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDC)等。作为工序G-2中使用的碱,可举出三乙胺、吡啶、N,N-二乙基苯胺、4-二甲基氨基吡啶、二异丙基乙胺等有机碱。作为上述工序(G-1),例如,可举出下述的工序G-1A~工序G-1F。工序G-1A:使环状膦酸化合物(8)与油酸在上述缩合剂的存在下反应的方法;工序G-1B:使环状膦酸化合物(8)与油酸在2-氯-1-甲基吡啶鎓碘化物(2-chloro-1-methylpyridiniumiodide,CMPI)的存在下反应的方法;工序G-1C:使环状膦酸化合物(8)与油酸在六氟磷酸(苯并三唑-1-基氧基)三吡咯烷基鏻((benzotriazol-1-yl-oxy)tripyrrolidinophosphoniumhexafluorophosphate,pyBOP)的存在下反应的方法;工序G-1D:使环状膦酸化合物(8)与油酸在O-(7-氮杂苯并三唑-1-基)-N,N,N,N-四甲基脲鎓六氟磷酸盐(O-(7-azabenzotriazol-1-yl)-N,N,N,N-tetramethyluroniumhexafluorophosphate,HATU)的存在下反应的方法;工序G-1E:使环状膦酸化合物(8)与油酸在O-(苯并三唑-1-基)-N,N,N,N-四甲基脲鎓六氟磷酸盐(O-(benzotriazol-1-yl)-N,N,N,N-tetramethyluroniumhexafluorophosphate,HBTU)的存在下反应的方法;工序G-1F:使环状膦酸化合物(8)与油酸在(1-氰基-2-乙氧基-2-氧代亚乙基氨基氧基)二甲基氨基-吗啉-碳鎓六氟磷酸盐(COMU)的存在下反应的方法。作为上述工序(G-2),例如,可举出下述的工序G-2A及工序G-2B。工序G-2A:使环状膦酸化合物(8)与油酰卤化物在三乙胺的存在下反应的方法;工序G-2B:由油酸制造油酰卤化物,使该油酰卤化物与环状膦酸化合物(8)在三乙胺的存在下反应的方法;作为上述工序G-3,例如,可举出下述的工序G-3A及工序G-3B。工序G-3A:使环状膦酸化合物(8)与油酸酐反应的方法;工序G-3B:使油酸与对甲苯磺酰氯反应,在反应体系中生成油酸的混合酸酐,然后使该酸酐与环状膦酸化合物(8)反应的方法对于缩合剂或碱而言,相对于1摩尔环状膦酸化合物(8),通常可以以从0.25摩尔至过量为止的任意比例进行使用,优选为0.5~2摩尔。这些缩合剂或碱可根据油酸化合物或其衍生物的种类而适宜选择。工序(G)可在氮、氩等非活性气体的气氛下进行。反应压力没有特别限制,可在常压下或加压下实施。反应温度通常为0~120℃,优选为0~30℃,更优选为15~25℃。反应时间通常为0.1~100小时,优选为0.5~50小时,更优选为2~17小时。反应结束后,可利用分液、浓缩、柱纯化等通常的分离方法将过量的试剂(油酸化合物等)、未反应的原料化合物等从得到的反应混合物中除去,并取出作为目标的通式(9)表示的化合物。此外,反应结束后,也可仅进行分液及浓缩、而不经过纯化及分离工序地将反应后的混合物直接用于工序(H)(叠缩合成)。实施例以下,给出合成例、实施例及比较例,对本发明进一步详细地进行说明,但本发明不受以下实施例的任何限定。[工序(A)]合成例A1(工序A:R2=正丙基)2,2-二正丙基-5-羟甲基-1,3-二氧杂环己烷(3b)的合成[化学式36]使3.0g的2-羟甲基-1,3-丙二醇(2)溶解于30ml四氢呋喃中,加入4.74ml的4-庚酮及53.8mg对甲苯磺酸一水合物,利用迪恩-斯塔克(Dean-Stark)装置加热回流3.5小时。此外,反应中,废弃被蒸馏除去的四氢呋喃,并向反应混合物中加入新的四氢呋喃。反应后,向反应混合物中加入0.39ml三乙胺,停止反应,在减压下蒸馏除去四氢呋喃。向残余物中加入30ml乙酸乙酯及30ml水,进行分液。萃取有机层后,进一步用30ml乙酸乙酯萃取水层2次。将收集的有机层用30ml饱和食盐水洗涤,然后用硫酸镁干燥,过滤,接着在减压下蒸馏除去乙酸乙酯。将得到的残余物利用硅胶柱色谱法(正己烷∶乙酸乙酯=1∶1)进行纯化,以41%的收率得到了2.34g缩醛化合物(3b)。1H-NMR(500MHz,CDCl3):δ:0.93(m,6H),1.35(m,4H),1.63(m,3H),1.72(m,2H),1.79(m,1H),3.76(m,4H),4.01(dd,J=11.9,4.0Hz,2H)合成例A2(工序A:R2=正丁基)2,2-二丁基-5-羟甲基-1,3-二氧杂环己烷(3c)的合成[化学式37]使1.0g的2-羟甲基-1,3-丙二醇(2)溶解于9.5ml四氢呋喃中,加入1.96ml的5-壬酮及17.9mg对甲苯磺酸一水合物,加热回流17小时。向反应混合物中加入0.13ml三乙胺,停止反应,在减压下蒸馏除去四氢呋喃。向残余物中加入10ml乙酸乙酯及10ml水,进行分液。萃取有机层后,进一步用10ml乙酸乙酯萃取水层2次。将收集的有机层用10ml饱和食盐水洗涤,然后用硫酸镁干燥,过滤,接着在减压下蒸馏除去乙酸乙酯。将得到的残余物利用硅胶柱色谱法(正己烷∶乙酸乙酯=1∶1)进行纯化,以7%的收率得到了156mg缩醛化合物(3c)。1H-NMR(500MHz,CDCl3):δ:0.92(m,6H),1.31(m,8H),1.50(t,J=5.1Hz,1H),1.65(m,2H),1.74(m,2H),1.80(m,1H),3.76(m,4H),4.01(dd,J=12.1,4.1Hz,2H)合成例A3(工序A:R2=苯基)2,2-二苯基-5-羟甲基-1,3-二氧杂环己烷(3d)的合成[化学式38]使1.0g的2-羟甲基-1,3-丙二醇(2)溶解于48ml的DMF中,加入2.58g苯甲酮二甲基缩酮及656mg的CSA,于40℃、减压下搅拌22.5小时。在减压下蒸馏除去反应混合物中的DMF,向残余物中加入10ml乙酸乙酯及10ml水,进行分液。萃取有机层后,进一步用10ml乙酸乙酯萃取水层2次。将收集的有机层用10ml饱和食盐水洗涤。用硫酸镁干燥,过滤,接着在减压下蒸馏除去乙酸乙酯。将得到的残余物利用硅胶柱色谱法(正己烷∶乙酸乙酯=1∶1)进行纯化,以25%的收率得到了647mg缩醛化合物(3d)。1H-NMR(500MHz,CDCl3)δ:1.94(m,1H),3.81(dd,J=7.0,5.2Hz,2H),3.91(dd,J=11.8,5.5Hz,2H),4.14(dd,J=11.8,3.8Hz,2H),7.25(m,2H),7.34(m,4H),7.51(m,4H)[工序(B)]合成例B1(工序B:R2=甲基)2,2-二甲基-5-甲磺酰氧基甲基-1,3-二氧杂环己烷(4a)的合成[化学式39]使5.00g的2,2-二甲基-5-羟甲基-1,3-二氧杂环己烷(3a)溶解于67ml的CH2Cl2中,接下来,加入5.42ml三乙胺,冷却至-20℃。进一步地,加入2.56ml甲磺酰氯(MsCl),于-20℃搅拌1小时。向反应混合物中加入50ml水,使反应停止,利用40ml的CH2Cl2萃取2次。将分液后得到的有机层用水洗涤,然后在减压下蒸馏除去CH2Cl2,将得到的残余物利用硅胶柱色谱法(正己烷∶乙酸乙酯=1∶1)进行纯化,以92%的收率得到了6.22g化合物(4a)。1H-NMR(500MHz,CDCl3):δ:1.39(s,3H),1.46(s,3H),2.00(m,1H),3.04(s,3H),3.78(dd,2H,J=12,3Hz),4.08(dd,2H,J=11.5,2.5Hz),4.42(d,2H,J=7Hz)合成例B2(工序B:R2=正丙基)2,2-二正丙基-5-甲磺酰氧基甲基-1,3-二氧杂环己烷(4b)的合成[化学式40]使1.0g合成例A1中得到的化合物(3b)溶解于20ml的CH2Cl2中,进一步加入1.03ml三乙胺,冷却至-20℃。接下来,向反应溶液中加入0.459ml的MsCl,于-20℃搅拌1小时。向得到的反应混合物中加入20ml水,使反应停止,用20ml的CH2Cl2萃取2次,用20ml饱和食盐水洗涤。接下来,用硫酸镁干燥,过滤,然后在减压下蒸馏除去CH2Cl2,将得到的残余物利用硅胶柱色谱法(正己烷∶乙酸乙酯=3∶1)进行纯化,以92%的收率得到了1.27g化合物(4b)。1H-NMR(500MHz,CDCl3)δ:0.93(m,6H),1.36(m,4H),1.58(m,1H),1.76(m,2H),1.95(m,1H),3.04(s,3H),3.74(dd,J=12.4,3.3Hz,2H),4.08(dd,J=15.6,3.3Hz,2H),4.43(d,J=7.5Hz,2H)合成例B3(工序B:R2=正丁基)2,2-二正丁基-5-甲磺酰氧基甲基-1,3-二氧杂环己烷(4c)的合成[化学式41]使156mg合成例A2中得到的化合物(3c)溶解于2.7ml的CH2Cl2中,进一步加入0.14ml三乙胺,冷却至-20℃。接下来,加入0.062ml的MsCl,于-20℃搅拌1小时。向得到的反应混合物中加入1.8ml水,使反应停止,用1.8ml的CH2Cl2萃取2次,将有机层用1.8ml饱和食盐水洗涤。接下来,用硫酸镁干燥,过滤,然后在减压下蒸馏除去CH2Cl2,将得到的残余物利用硅胶柱色谱法(正己烷∶乙酸乙酯=3∶1)进行纯化,以81%的收率得到了169mg化合物(4c)。1H-NMR(500MHz,CDCl3)δ:0.92(m,6H),1.30(m,8H),1.61(m,2H),1.78(m,2H),1.96(m,1H),3.04(s,3H),3.74(dd,J=12.6,3.6Hz,2H),4.08(dd,J=12.5,3.5Hz,2H),4.4-3(d,J=7.4Hz,2H)合成例B4(工序B:R2=苯基)2,2-二苯基-5-甲磺酰氧基甲基-1,3-二氧杂环己烷(4d)的合成[化学式42]使640mg合成例A3中得到的化合物(3d)溶解于9.6ml的CH2Cl2中,进一步加入493μl三乙胺,冷却至-20℃。接着,加入220μl的MsCl,于-20℃搅拌1小时。向得到的反应混合物加入6.4ml水,使反应停止,用6.4ml的CH2Cl2萃取2次,将有机层用6.4ml饱和食盐水洗涤。接着用硫酸镁干燥,过滤,然后在减压下蒸馏除去CH2Cl2,将得到的残余物利用硅胶柱色谱法(正己烷∶乙酸乙酯=3∶1)进行纯化,以88%的收率得到了729mg化合物(4d)。1H-NMR(500MHz,CDCl3)δ:2.06(m,1H),3.03(s,3H),3.95(dd,J=12.2,3.7Hz,2H),4.18(dd,J=12.2,3.4Hz,2H),4.51(d,J=7.5Hz,2H),7.25(m,1H),7.30(m,3H),7.39(m,2H),7.49(m,4H)[工序(C)]合成例C1-1(工序C:R2=甲基,碱=TEA,溶剂=甲基乙基酮)2,2-二甲基-5-碘甲基-1,3-二氧杂环己烷(5a)的合成[化学式43]使400.0mg合成例B1中得到的化合物(4a)溶解于6ml甲基乙基酮中,向其中加入12μl三乙胺及401.1mg碘化钠,加热回流1.5小时。接下来,在减压下蒸馏除去反应混合物中的甲基乙基酮,向其中加入10ml的CH2Cl2及10ml水,进行分液。将水层用10ml的CH2Cl2萃取2次,加入5ml的5%硫代硫酸钠及5ml的1%碳酸氢钠水溶液对有机层进行洗涤。进一步地,用10ml水对有机层进行洗涤,在减压下蒸馏除去CH2Cl2。将得到的残余物利用硅胶柱色谱法(正己烷∶乙酸乙酯=5∶1)进行纯化,以86%的收率得到了394.1g化合物(5a)。1H-NMR(500MHz,CDCl3)δ:1.41(s,3H),1.43(s,3H),1.95(m,1H),3.23(d,J=7Hz,2H),3.73(dd,J=12,6.5Hz,2H),4.01(dd,J=11.5,4Hz,2H)合成例C1-2(工序C:R2=正丙基)2,2-二正丙基-5-碘甲基-1,3-二氧杂环己烷(5b)的合成[化学式44]使1.20g合成例B2中得到的化合物(4b)溶解于14.3ml甲基乙基酮中,进而加入30μl三乙胺及966mg碘化钠,加热回流2小时。在减压下蒸馏除去反应液中的甲基乙基酮,加入15ml的CH2Cl2及15ml水,进行分液。将水层用15ml的CH2Cl2萃取2次,将有机层用7.5ml的5%硫代硫酸钠及7.5ml的1%碳酸氢钠水溶液洗涤。进一步地,用15ml饱和食盐水对有机层进行洗涤。接下来,用硫酸镁干燥,过滤,然后在减压下蒸馏除去CH2Cl2,将得到的残余物利用硅胶柱色谱法(正己烷∶乙酸乙酯=10∶1)进行纯化,以86%的收率得到了1.16g化合物(5b)。1H-NMR(500MHz,CDCl3)δ:0.93(t,J=7.3Hz,6H),1.35(m,4H),1.66(m,4H),1.90(m,1H),3.25(d,J=7.2Hz,2H),3.71(dd,J=11.8,6.0Hz,2H),4.01(dd,J=11.9,3.9Hz,2H)合成例C1-3(工序C:R2=正丁基)2,2-二正丁基-5-碘甲基-1,3-二氧杂环己烷(5c)的合成[化学式45]使169mg合成例B3中得到的化合物(4c)溶解于2.9ml甲基乙基酮中,进而加入3.8μl三乙胺及123mg碘化钠,加热回流2小时。接下来,在减压下蒸馏除去反应液中的甲基乙基酮,加入3ml的CH2Cl2及3ml水,进行分液。接下来,将水层用3ml的CH2Cl2萃取2次,将有机层用1.5ml的5%硫代硫酸钠及1.5ml的1%碳酸氢钠水溶液洗涤。进一步地,用3ml饱和食盐水对有机层进行洗涤。接下来,用硫酸镁干燥,过滤,然后在减压下蒸馏除去CH2Cl2。将得到的残余物利用硅胶柱色谱法(正己烷∶乙酸乙酯=10∶1)进行纯化,以75%的收率得到了139mg化合物(5c)。1H-NMR(500MHz,CDCl3)δ:0.92(m,6H),1.30(m,8H),1.68(m,4H),1.91(m,1H),3.25(d,J=7.2Hz,2H),3.71(dd,J=12.0,6.2Hz,2H),4.00(dd,J=11.9,4.0Hz,2H)合成例C1-4(工序C:R2=苯基)2,2-二苯基-5-碘甲基-1,3-二氧杂环己烷(5d)的合成[化学式46]使729mg合成例B4中得到的化合物(4d)溶解于10.9ml甲基乙基酮中,进而加入14.5μl三乙胺及470mg碘化钠,加热回流2小时。在减压下蒸馏除去反应液中的甲基乙基酮,加入5ml的CH2Cl2及5ml水,进行分液。将水层用3ml的CH2Cl2萃取2次,将有机层用2.5ml的5%硫代硫酸钠及2.5ml的1%碳酸氢钠水溶液洗涤。进一步地,用5ml饱和食盐水洗涤有机层。接下来,用硫酸镁干燥,过滤,然后在减压下蒸馏除去CH2Cl2。将得到的残余物利用硅胶柱色谱法(正己烷∶乙酸乙酯=10∶1)进行纯化,以91%的收率得到了726mg化合物(5d)。1H-NMR(500MHz,CDCl3)δ:2.10(m,1H),3.25(d,J=7.5Hz,2H),3.84(dd,J=11.7,6.3Hz,2H),4.16(dd,J=11.7,3.8Hz,2H),7.26(m,2H),7.34(m,4H),7.50(m,4H)合成例C2-1(工序C:溶剂=丙酮+甲基异丁基酮)2,2-二甲基-5-碘甲基-1,3-二氧杂环己烷(5a)的合成[化学式47]使300mg合成例B1中得到的化合物(4a)在氩气气氛下溶解于3ml丙酮及3ml甲基异丁基酮中,进而加入9.3μl三乙胺及301mg碘化钠,加热回流2小时。在减压下蒸馏除去反应液中的丙酮及甲基异丁基酮,向其中加入6ml的CH2Cl2及6ml水,进行分液。将水层用6ml的CH2Cl2萃取2次,将有机层用3ml的5%硫代硫酸钠及3ml的1%碳酸氢钠水溶液洗涤。进一步地,用6ml饱和食盐水对有机层进行洗涤,并用硫酸镁干燥,然后在减压下蒸馏除去CH2Cl2。将得到的残余物利用硅胶柱色谱法(正己烷:乙酸乙酯=10:1)进行纯化,以78%的收率得到了266mg化合物(5a)。合成例C2-2(工序C:溶剂=丙酮)2,2-二甲基-5-碘甲基-1,3-二氧杂环己烷(5a)的合成[化学式48]使300mg合成例B1中得到的化合物(4a)在氩气氛下溶解于6ml丙酮中,进而加入9.3μl三乙胺及301mg碘化钠,加热回流2小时。在减压下蒸馏除去反应液中的丙酮,加入6ml的CH2Cl2及6ml水,进行分液。将水层用6ml的CH2Cl2萃取2次,并将有机层用3ml的5%硫代硫酸钠及3ml的1%碳酸氢钠水溶液洗涤。进一步地,用6ml饱和食盐水对有机层进行洗涤,并用硫酸镁干燥,然后在减压下蒸馏除去CH2Cl2。将得到的残余物利用硅胶柱色谱法(正己烷:乙酸乙酯=10:1)进行纯化,以59%的收率得到了204mg化合物(5a)。合成例C2-3(工序C:溶剂=甲基异丁基酮)2,2-二甲基-5-碘甲基-1,3-二氧杂环己烷(5a)的合成[化学式49]使300mg合成例B1中得到的化合物(4a)在氩气气氛下溶解于6ml甲基异丁基酮中,加入9.3μl三乙胺及301mg碘化钠,加热回流2小时。在减压下蒸馏除去反应液中的甲基异丁基酮,加入6mlCH2Cl2及6ml水,进行分液。将水层用6ml的CH2Cl2萃取2次,并将有机层用3ml的5%硫代硫酸钠及3ml的1%碳酸氢钠水溶液洗涤。进一步地,用6ml饱和食盐水对有机层进行洗涤,并用硫酸镁干燥,然后在减压下蒸馏除去CH2Cl2。将得到的残余物利用硅胶柱色谱法(正己烷:乙酸乙酯=10:1)进行纯化,以55%的收率得到了190mg化合物(5a)。合成例C3-1(工序C:碱金属卤化物=溴化钠)2,2-二甲基-5-溴甲基-1,3-二氧杂环己烷(5a-1)的合成[化学式50]使300mg合成例B1中得到的化合物(4a)在氩气气氛下溶解于6ml甲基乙基酮中,进而加入9.3μl三乙胺及207mg溴化钠,加热回流23小时。在减压下蒸馏除去反应液中的甲基乙基酮,加入6ml的CH2Cl2及6ml水进行分液。将水层用6ml的CH2Cl2萃取2次,并将有机层用3ml的5%硫代硫酸钠及3ml的1%碳酸氢钠水溶液洗涤。进一步地,用6ml饱和食盐水对有机层进行洗涤,并用硫酸镁干燥,然后在减压下蒸馏除去CH2Cl2。将得到的残余物利用硅胶柱色谱法(正己烷:乙酸乙酯=10:1)进行纯化,以45%的收率得到了127mg化合物(5a-1)。1H-NMR(500MHz,CDCl3)δ:1.41(s,3H),1.44(s,3H),2.02(m,1H),3.51(d,J=7.1Hz,2H),3.80(dd,J=12,5.7Hz,2H),4.05(dd,J=12,4Hz,2H)合成例C4-1(工序C:碱=二异丙基乙胺)2,2-二甲基-5-碘甲基-1,3-二氧杂环己烷(5a)的合成[化学式51]使300mg合成例B1中得到的化合物(4a)在氩气气氛下溶解于6ml甲基乙基酮中,进而加入12μl二异丙基乙胺及301mg碘化钠,加热回流2小时。在减压下蒸馏除去反应液中的甲基乙基酮,加入6ml的CH2Cl2及6ml水,进行分液。将水层用6ml的CH2Cl2萃取2次,并将有机层用3ml的5%硫代硫酸钠及3ml的1%碳酸氢钠水溶液洗涤。进一步地,用6ml饱和食盐水对有机层进行洗涤,并用硫酸镁干燥,然后在减压下蒸馏除去CH2Cl2。将得到的残余物利用硅胶柱色谱法(正己烷∶乙酸乙酯=10∶1)进行纯化,以78%的收率得到了262mg化合物(5a)。合成例C4-2(工序C:碱=碳酸氢钠,溶剂=甲基乙基酮)2,2-二甲基-5-碘甲基-1,3-二氧杂环己烷(5a)的合成[化学式52]使300mg合成例B1中得到的化合物(4a)在氩气气氛下溶解于6ml甲基乙基酮中,进而加入5.6mg碳酸氢钠及301mg碘化钠,加热回流2小时。在减压下蒸馏除去反应液中的甲基乙基酮,加入6ml的CH2Cl2及6ml水,进行分液。将水层用6ml的CH2Cl2萃取2次,并将有机层用3ml的5%硫代硫酸钠及3ml的1%碳酸氢钠水溶液洗涤。进一步地,用6ml饱和食盐水对有机层进行洗涤,并用硫酸镁干燥,然后在减压下蒸馏除去CH2Cl2。将得到的残余物利用硅胶柱色谱法(正己烷:乙酸乙酯=10:1)进行纯化,以68%的收率得到了234mg化合物(5a)。合成例C4-3(工序C:碱=NaHCO3,溶剂=丙酮)2,2-二甲基-5-碘甲基-1,3-二氧杂环己烷(5a)的合成[化学式53]使15.0g合成例B1中得到的化合物(4a)溶解于225ml丙酮中,向其中加入5.62g碳酸氢钠及20.05g碘化钠,加热回流19小时。接下来,过滤白色固体,并用丙酮洗涤,然后在减压下蒸馏除去丙酮,向其中加入150ml的CH2Cl2,向有机层中加入50ml的5%硫代硫酸钠及50ml的1%碳酸氢钠水溶液,进行分液。用50ml的CH2Cl2萃取,并用5%食盐水洗涤有机层,在减压下蒸馏除去CH2Cl2。将得到的残余物利用硅胶柱色谱法(正己烷:乙酸乙酯=5:1)进行纯化,以82%的收率得到了14.00g化合物(5a)。合成例C4-4(工序C:碱=碳酸氢钾)2,2-二甲基-5-碘甲基-1,3-二氧杂环己烷(5a)的合成[化学式54]使300mg合成例B1中得到的化合物(4a)在氩气气氛下溶解于6ml甲基乙基酮中,进而加入6.7mg碳酸氢钾及301mg碘化钠,加热回流2小时。在减压下蒸馏除去反应液中的甲基乙基酮,加入6ml的CH2Cl2及6ml水,进行分液。将水层用6ml的CH2Cl2萃取2次,并将有机层用3ml的5%硫代硫酸钠及3ml的1%碳酸氢钠水溶液洗涤。进一步地,用6ml饱和食盐水对有机层进行洗涤,并用硫酸镁干燥,然后,在减压下蒸馏除去CH2Cl2。将得到的残余物利用硅胶柱色谱法(正己烷:乙酸乙酯=10:1)进行纯化,以31%的收率得到了106mg化合物(5a)。合成例C4-5(工序C:碱=碳酸钾)2,2-二甲基-5-碘甲基-1,3-二氧杂环己烷(5a)的合成[化学式55]使300mg合成例B1中得到的化合物(4a)在氩气气氛下溶解于6ml甲基乙基酮中,进而加入301mg碳酸钾及碘化钠,加热回流2小时。在减压下蒸馏除去反应液中的甲基乙基酮,加入6ml的CH2Cl2及6ml水,进行分液。将水层用6ml的CH2Cl2萃取2次,并将有机层用3ml的5%硫代硫酸钠及3ml的1%碳酸氢钠水溶液洗涤。进一步地,用6ml饱和食盐水对有机层进行洗涤,并用硫酸镁干燥,然后在减压下蒸馏除去CH2Cl2。将得到的残余物利用硅胶柱色谱法(正己烷:乙酸乙酯=10:1)进行纯化,以61%的收率得到了209mg化合物(5a)。合成例C4-6(工序C:碱=碳酸钠)2,2-二甲基-5-碘甲基-1,3-二氧杂环己烷(5a)的合成[化学式56]使300mg合成例B1中得到的化合物(4a)在氩气气氛下溶解于6ml甲基乙基酮中,进而加入碳酸钠及301mg碘化钠,加热回流2小时。在减压下蒸馏除去反应液中的甲基乙基酮,加入6ml的CH2Cl2及6ml水,进行分液。将水层用6ml的CH2Cl2萃取2次,并将有机层用3ml的5%硫代硫酸钠及3ml的1%碳酸氢钠水溶液洗涤。进一步地,用6ml饱和食盐水洗涤有机层,并用硫酸镁干燥,然后在减压下蒸馏除去CH2Cl2。将得到的残余物利用硅胶柱色谱法(正己烷:乙酸乙酯=10:1)进行纯化,以79%的收率得到了270mg化合物(5a)。[工序(B’)]合成例B1(工序B’:溶剂=二氯甲烷)2,2-二甲基-5-碘甲基-1,3-二氧杂环己烷(5a)的合成[化学式57]使1.00g的2,2-二甲基-5-羟甲基-1,3-二氧杂环己烷(3a)溶解于22.8ml二氯甲烷(MDC)中,向其中加入700.0mg咪唑及2.08g碘,冷却至0℃后,接着加入2.15g三苯基膦,于25℃搅拌2小时。然后,用20ml水进行淬灭,用10ml二氯甲烷萃取2次,并用20ml水洗涤。在减压下蒸馏除去有机相中的二氯甲烷,将得到的残余物利用硅胶柱色谱法(正己烷:乙酸乙酯=5:1)进行纯化,以79%的收率得到了1.38g的2,2-二甲基-5-碘甲基-1,3-二氧杂环己烷(5a)。合成例B2(工序B’:溶剂=THF)2,2-二甲基-5-碘甲基-1,3-二氧杂环己烷(5a)的合成[化学式58]使500.0mg的2,2-二甲基-5-羟甲基-1,3-二氧杂环己烷(3a)溶解于3.4ml四氢呋喃中,冷却至0℃,然后向其中分别加入1.08g三苯基膦、279.5mg咪唑、及1.04g碘,于25℃搅拌2.5小时。然后,用10ml的5%硫代硫酸钠水溶液洗涤,用20ml乙酸乙酯萃取,并用5%食盐水洗涤。将有机相用硫酸镁干燥后,过滤,在减压下蒸馏除去乙酸乙酯,将得到的残余物利用硅胶柱色谱法(正己烷:乙酸乙酯=5:1)进行纯化,以73%的收率得到了639.0mg的2,2-二甲基-5-碘甲基-1,3-二氧杂环己烷(5a)。合成例B3(工序B’:溶剂=CPME)2,2-二甲基-5-溴甲基-1,3-二氧杂环己烷(5a)的合成[化学式59]使500.0mg的2,2-二甲基-5-羟甲基-1,3-二氧杂环己烷(3a)溶解于6.8ml四氢呋喃中,冷却至0℃,然后分别加入1.08g三苯基膦、279.5mg咪唑、及1.04g碘,并于25℃搅拌2小时。然后,除去三苯基氧化膦,并用10ml的CPME洗涤,然后将有机相用10ml的5%硫代硫酸钠水溶液洗涤,并用10ml的CPME萃取3次,用5%食盐水洗涤。在减压下蒸馏除去CPME,将得到的残余物利用硅胶柱色谱法(正己烷:乙酸乙酯=5:1)进行纯化,以71%的收率得到了621.1mg的2,2-二甲基-5-溴甲基-1,3-二氧杂环己烷(5a)。合成例B4(工序B’:溶剂=二氯甲烷)2,2-二甲基-5-溴甲基-1,3-二氧杂环己烷(5b)的合成[化学式60]使1.00g的2,2-二甲基-5-羟甲基-1,3-二氧杂环己烷(3a)溶解于22.8ml二氯甲烷(MDC)中,向其中加入700.0mg咪唑、及2.72g四溴化碳,进行冰冷却后,接着加入2.15g三苯基膦,于室温搅拌4小时。在减压下蒸馏除去二氯甲烷,利用硅胶柱色谱法(正己烷:乙酸乙酯=5:1)进行纯化,定量地得到了1.60g的2,2-二甲基-5-溴甲基-1,3-二氧杂环己烷(5b)。[工序(D)]合成例D1-1(工序D:R2=甲基)(2,2-二甲基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二甲酯(6a)的合成[化学式61]使300.0mg合成例C1-1中得到的碘化合物(5a)溶解于2.34ml的DMF中,进而加入763.4mg碳酸铯及215μl亚磷酸二甲酯,于40℃搅拌5小时。在减压下蒸馏除去反应液中的DMF,向得到的残余物中加入5ml甲苯进行置换,过滤白色固体。进而将该白色固体用5ml甲苯洗涤,然后将得到的滤液在减压下浓缩,并将残余物利用硅胶柱色谱法(氯仿∶甲醇=10∶1)进行纯化,以92%的收率得到了255.6mg膦酸二甲酯化合物(6a)。1H-NMR(500MHz,CDCl3)δ:1.42(s,6H),1.82(dd,J=18.5,6.5Hz,2H),2.15(m,1H),3.66(dd,J=11.5,7Hz,2H),3.75(d,J=10.5Hz,6H),4.01(d,J=12,3.5Hz,2H)合成例D1-2(工序D:R2=正丙基)(2,2-二正丙基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二甲酯(6b)的合成[化学式62]使1.06g合成例C1-2中得到的碘化合物(5b)溶解于6.8ml的DMF中,进而加入2.21g碳酸铯及621μl亚磷酸二甲酯,于40℃搅拌3小时。在减压下蒸馏除去反应液中的DMF,向得到的残余物中加入5ml甲苯进行置换,过滤白色固体。进而将该白色固体用2.5ml甲苯洗涤,然后将得到的滤液在减压下浓缩,并将残余物利用硅胶柱色谱法(氯仿∶甲醇=20∶1)进行纯化,以79%的收率得到了767mg膦酸二甲酯化合物(6b)。1H-NMR(500MHz,CDCl3)δ:0.91(t,J=7.4Hz,6H),1.36(m,4H),1.66(m,4H),1.84(dd,J=18.7,6.9Hz,2H),2.09(m,1H),3.63(dd,J=11.7,6.6Hz,2H),3.75(d,J=10.9Hz,6H),3.99(dd、J=11.7,3.8Hz,2H)合成例D1-3(工序D:R2=丁基)(2,2-二丁基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二甲酯(6c)的合成[化学式63]使139mg合成例C1-3中得到的碘化合物(5c)溶解于1.2ml的DMF中,进而加入266mg碳酸铯及75μl亚磷酸二甲酯,于50℃搅拌3小时。在减压下蒸馏除去反应液中的DMF,向得到的残余物中加入1ml甲苯进行置换,过滤白色固体。进而将该白色固体用1ml甲苯洗涤,然后将得到的滤液在减压下浓缩,并将残余物利用硅胶柱色谱法(氯仿∶甲醇=10∶1)进行纯化,以61%的收率得到了80.5mg膦酸二甲酯化合物(6c)。1H-NMR(500MHz,CDCl3)δ:0.91(m,6H),1.30(m,8H),1.68(m,4H),1.83(dd,J=18.7,6.9Hz,2H),2.10(m,1H),3.63(dd.J=11.8,6.7Hz,2H),3.75(d,J=10.9Hz,6H),3.99(dd,J=11.7,3.9Hz,2H)合成例D1-4(工序D:R2=苯基)(2,2-二苯基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二甲酯(6d)的合成[化学式64]使726mg合成例C1-4中得到的碘化合物(5d)溶解于5.7ml的DMF中,进而加入1.24g碳酸铯及350μl亚磷酸二甲酯,于50℃搅拌3小时。在减压下蒸馏除去反应液中的DMF,向得到的残余物中加入4.2ml甲苯进行置换,过滤白色固体。进而将该白色固体用2.1ml甲苯洗涤,然后将得到的滤液在减压下浓缩,并将残余物利用硅胶柱色谱法(氯仿∶甲醇=10∶1)进行纯化,以90%的收率得到了621mg膦酸二甲酯化合物(6d)。1H-NMR(500MHz,CDCl3)δ:1.80(dd,J=18.8,7.0Hz,2H),2.30(m,1H),3.74(d,J=10.9Hz,6H),3.77(dd,J=11.5,7.2Hz,2H),4.15(dd,J=11.5,3.8Hz,2H),7.26(m,2H),7.33(m,4H),7.50(m,4H)合成例D2-1(工序D:R2=甲基,溶剂=DMF)(2,2-二甲基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二甲酯(6a)的合成[化学式65]使500.0mg合成例C1-1中得到的碘化合物(5a)溶解于3.9ml的DMF中,进而加入1.27g碳酸铯及360μl亚磷酸二甲酯,于50℃搅拌3小时。在减压下蒸馏除去反应液中的DMF,向得到的残余物中加入10ml甲苯进行置换,过滤白色固体。进而将该白色固体用10ml甲苯洗涤,然后将得到的滤液在减压下浓缩,并将残余物利用硅胶柱色谱法(氯仿:甲醇=10:1)进行纯化,以87%的收率得到了406.0mg膦酸二甲酯化合物(6a)。合成例D2-2(工序D:R2=甲基,溶剂=DMAc)(2,2-二甲基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二甲酯(6a)的合成[化学式66]使500.0mg合成例C1-1中得到的碘化合物(5a)溶解于3.9ml的DMAc中,进而加入1.27g碳酸铯及360μl亚磷酸二甲酯,于50℃搅拌3小时。在减压下蒸馏除去反应液中的DMAc,向得到的残余物中加入10ml甲苯进行置换,过滤白色固体。进而将该白色固体用10ml甲苯洗涤,然后将得到的滤液在减压下浓缩,并将残余物利用硅胶柱色谱法(氯仿:甲醇=10:1)进行纯化,以94%的收率得到了438.7mg膦酸二甲酯化合物(6a)。合成例D2-3(工序D:R2=甲基,溶剂=乙腈)(2,2-二甲基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二甲酯(6a)的合成[化学式67]使500.0mg合成例C1-1中得到的碘化合物(5a)溶解于3.9ml乙腈中,进而加入1.27g碳酸铯及360μl亚磷酸二甲酯,于50℃搅拌24小时。在减压下蒸馏除去反应液中的乙腈,向得到的残余物中加入20ml甲苯进行置换,过滤白色固体。进而将该白色固体用10ml甲苯洗涤,然后将得到的滤液在减压下浓缩,并将残余物利用硅胶柱色谱法(氯仿:甲醇=10:1)进行纯化,以88%的收率得到了408.8mg膦酸二甲酯化合物(6a)。合成例D2-4(工序D:R2=甲基,溶剂=DMF/AN(1/1))(2,2-二甲基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二甲酯(6a)的合成[化学式68]使500.0mg合成例C1-1中得到的碘化合物(5a)溶解于2.0ml的DMF、2.0ml乙腈中,进而加入1.27g碳酸铯及360μl亚磷酸二甲酯,于50℃搅拌18小时。在减压下蒸馏除去反应液中的溶剂,向得到的残余物中加入20ml甲苯进行置换,过滤白色固体。进而将该白色固体用10ml甲苯洗涤,然后将得到的滤液在减压下浓缩,并将残余物利用硅胶柱色谱法(氯仿:甲醇=10:1)进行纯化,以87%的收率得到了404.1mg膦酸二甲酯化合物(6a)。合成例D3-1(工序D:R2=甲基,碱=碳酸铯)(2,2-二甲基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二甲酯(6a)的合成[化学式69]使500.0mg合成例C1-1中得到的碘化合物(5a)溶解于3.9ml的DMF中,进而加入1.27g碳酸铯及360μl亚磷酸二甲酯,于50℃搅拌3小时。在减压下蒸馏除去反应液中的DMF,向得到的残余物中加入10ml甲苯进行置换,过滤白色固体。进而将该白色固体用10ml甲苯洗涤,然后将得到的滤液在减压下浓缩,并将残余物利用硅胶柱色谱法(氯仿:甲醇=10:1)进行纯化,以87%的收率得到了406.0mg膦酸二甲酯化合物(6a)。合成例D3-2(工序D:R2=甲基,碱=碳酸钾)(2,2-二甲基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二甲酯(6a)的合成[化学式70]使500.0mg合成例C1-1中得到的碘化合物(5a)溶解于3.9ml的DMF中,进而加入540mg碳酸钾及360μl亚磷酸二甲酯,于50℃搅拌24小时。在减压下蒸馏除去反应液中的DMF,向得到的残余物中加入10ml甲苯进行置换,过滤白色固体。进而将该白色固体用10ml甲苯洗涤,然后将得到的滤液在减压下浓缩,并将残余物利用硅胶柱色谱法(氯仿:甲醇=10:1)进行纯化,以30%的收率得到了139.6mg膦酸二甲酯化合物(6a)。合成例D3-3(工序D:R2=甲基,碱=碳酸铷)(2,2-二甲基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二甲酯(6a)的合成[化学式71]使500.0mg合成例C1-1中得到的碘化合物(5a)溶解于3.9ml的DMF中,进而加入902.1mg碳酸铷及360μl亚磷酸二甲酯,于50℃搅拌24小时。在减压下蒸馏除去反应液中的DMF,向得到的残余物中加入10ml甲苯进行置换,过滤白色固体。进而将该白色固体用10ml甲苯洗涤,然后将得到的滤液在减压下浓缩,并将残余物利用硅胶柱色谱法(氯仿:甲醇=10:1)进行纯化,以60%的收率得到了280.7mg膦酸二甲酯化合物(6a)。合成例D3-4(工序D:R2=甲基,碱=碳酸钠)(2,2-二甲基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二甲酯(6a)的合成[化学式72]使500.0mg合成例C1-1中得到的碘化合物(5a)溶解于3.9ml的DMF中,进而加入414.0mg碳酸钠及360μl亚磷酸二甲酯,于50℃搅拌24小时。在减压下蒸馏除去反应液中的DMF,向得到的残余物中加入10ml甲苯进行置换,过滤白色固体。进而将该白色固体用10ml甲苯洗涤,然后将得到的滤液在减压下浓缩,将残余物用甲苯定容至5ml,进行液体的定量,测得收率为1.2%。合成例D3-5(工序D:R2=甲基,碱=碳酸氢钾)(2,2-二甲基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二甲酯(6a)的合成[化学式73]使500.0mg合成例C1-1中得到的碘化合物(5a)溶解于3.9ml的DMF中,进而加入391.1mg碳酸氢钾及360μl亚磷酸二甲酯,于50℃搅拌24小时。在减压下蒸馏除去反应液中的DMF,向得到的残余物中加入10ml甲苯进行置换,过滤白色固体。进而将该白色固体用10ml甲苯洗涤,然后将得到的滤液在减压下浓缩,将残余物用甲苯定容至5ml,进行液体的定量,测得收率为3.5%。合成例D4-1(工序D:X=溴)(2,2-二甲基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二甲酯(6a)的合成[化学式74]使93.7mg合成例C3-1中得到的溴化合物(5a-1)溶解于0.9ml的DMF中,进而加入292mg碳酸铯及82.1μl亚磷酸二甲酯,于50℃搅拌4小时。在减压下蒸馏除去反应液中的DMF,向得到的残余物中加入5ml甲苯进行置换,过滤白色固体。进而将该白色固体用10ml甲苯洗涤,然后将得到的滤液在减压下浓缩,并将残余物利用硅胶柱色谱法(氯仿:甲醇=10:1)进行纯化,以84%的收率得到了89.3mg膦酸二甲酯化合物(6a)。合成例D5-1(工序D:R1=乙基)(2,2-二甲基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二乙酯(6a-1)的合成[化学式75]使2.00g合成例C1-1中得到的碘化合物(5a)溶解于15.6ml的DMF中,进而加入5.09g碳酸铯及2.01ml亚磷酸二乙酯,于50℃搅拌3小时。在减压下蒸馏除去反应液中的DMF,向得到的残余物中加入20ml甲苯进行置换,过滤白色固体。进而将该白色固体用30ml甲苯洗涤,然后将得到的滤液在减压下浓缩,并将残余物利用硅胶柱色谱法(氯仿∶甲醇=10∶1)进行纯化,定量地得到了2.24g膦酸二乙酯化合物(6a-1)。1H-NMR(500MHz,CDCl3)δ:1.33(t,J=7Hz,6H),1.42(s,3H),1.42(s,3H),1.76(dd,J=19,7Hz,2H),2.18(m,1H),3.66(dd,J=11.5,7.5Hz,2H),4.00(dd,J=11.5,4Hz,2H),4.10(m,4H)合成例D5-2(工序D:R1=正丁基)(2,2-二甲基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二丁酯(6a-2)的合成[化学式76]使2.00g合成例C1-1中得到的碘化合物(5a)溶解于15.6ml的DMF中,进而加入5.09g碳酸铯及3.05ml亚磷酸二丁酯,于50℃搅拌3小时。在减压下蒸馏除去反应液中的DMF,向得到的残余物中加入20ml甲苯进行置换,过滤白色固体。进而将该白色固体用20ml甲苯洗涤,然后将得到的滤液在减压下浓缩,将残余物利用硅胶柱色谱法(氯仿∶甲醇=10∶1)进行纯化,定量地得到了2.74g膦酸二丁酯化合物(6a-2)。1H-NMR(500MHz,CDCl3)δ:0.94(t,J=7.5Hz,6H),1.40(tq,J=7.5,7.5Hz,4H),1.42(s,6H),1.65(tt,J=8,8Hz,4H),1.75(dd,J=19,7Hz,2H),2.17(m,1H),3.66(dd,J=11.5,7.5Hz,2H),4.03(m,6H)合成例D5-3(工序D:R1=苄基)(2,2-二甲基-[1,3]二氧杂环己烷-5-基甲基)-膦酸二苄酯(6a-3)的合成[化学式77]使1.00g合成例C1-1中得到的碘化合物(5a)溶解于7.8ml的DMF中,进而加入2.54g碳酸铯及1.74ml亚磷酸二苄酯,于50℃搅拌3小时。在减压下蒸馏除去反应液中的DMF,向得到的残余物中加入10ml甲苯进行置换,过滤白色固体。进而将该白色固体用20ml甲苯洗涤,然后将得到的滤液在减压下浓缩,并将残余物利用硅胶柱色谱法(正己烷∶乙酸乙酯=1∶2)进行纯化,定量地得到了1.58g膦酸二苄酯化合物(6a-3)。1H-NMR(500MHz,CDCl3)δ:1.38(s,3H),1.39(s,3H),1.77(dd,J=19,7Hz,2H),2.11(m,1H),3.58(dd,J=11.5,7Hz,2H),3.93(dd,12,4Hz,2H),5.00(m,4H),7.34(m,10H)[工序(E)]合成例E1-1(工序E:R2=甲基)(2,3-二羟基丙基)-膦酸二甲酯(7a)的合成[化学式78]使5.00g合成例D1-1中得到的膦酸二甲酯化合物(6a)溶解于125ml甲醇中,进而加入798.5mg对甲苯磺酸一水合物,于20℃搅拌3小时。用640μl三乙胺使反应停止,在减压下蒸馏除去反应液中的甲醇。将得到的残余物利用硅胶柱色谱法(氯仿∶甲醇=10∶1)进行纯化,以92%的收率得到了3.82g二元醇化合物(7a)。1H-NMR(500MHz,CDCl3)δ:1.93(dd,J=18.5,7Hz,2H),2.13(m,1H),2.87(dd,J=6,6Hz,2H),3.77(m,10H)合成例E1-2(工序E:R2=正丙基)2-(二甲基膦酰基)甲基-1,3-丙二醇(7a)的合成[化学式79]使687mg合成例D1-2中得到的膦酸二甲酯化合物(6b)溶解于4.7ml甲醇中,进而加入22.0mg对甲苯磺酸一水合物,于20℃搅拌3小时。在减压下蒸馏除去反应液中的甲醇。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=5:1)进行纯化,以78%的收率得到了361mg二元醇化合物(7a)。合成例E1-3(工序E:R2=丁基)2-(二甲基膦酰基)甲基-1,3-丙二醇(7a)的合成[化学式80]使80.5mg合成例D1-3中得到的膦酸二甲酯化合物(6c)溶解于0.75ml甲醇中,进而加入2.4mg对甲苯磺酸一水合物,于20℃搅拌3小时。在减压下蒸馏除去反应液中的甲醇。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=10:1)进行纯化,以76%的收率得到了37.5mg二元醇化合物(7a)。合成例E1-4(工序E:R2=苯基)2-(二甲基膦酰基)甲基-1,3-丙二醇(7a)的合成[化学式81]使621mg合成例D1-4中得到的膦酸二甲酯化合物(6d)溶解于5.1ml甲醇中,进而加入16.3mg对甲苯磺酸一水合物,于0℃搅拌15小时,继而于20℃搅拌1小时。在减压下蒸馏除去反应液中的甲醇。将得到的残余物利用硅胶柱色谱法(氯仿∶甲醇=10∶1)进行纯化,以67%的收率得到了226mg二元醇化合物(7a)。合成例E2-1(工序E:R2=乙基)2-(二乙基膦酰基)甲基-1,3-丙二醇(7a-1)的合成[化学式82]使2.00g合成例D5-1中得到的膦酸二乙酯化合物(6a-1)溶解于15ml甲醇中,进而加入428.6mg对甲苯磺酸一水合物,于20℃搅拌3小时,然后,于0℃搅拌过夜。在减压下蒸馏除去反应液中的甲醇,将得到的残余物利用硅胶柱色谱法(氯仿∶甲醇=5∶1)进行纯化,以99%的收率得到了1.69g二元醇化合物(7a-1)。1H-NMR(500MHz,CDCl3)δ:1.34(t,J=7Hz,6H),1.91(dd,J=18.5,7Hz,2H),2.14(m,1H),3.20(br,羟基),3.77(d,J=5Hz,4H),4.12(m,4H)合成例E2-2(工序E:R2=正丁基)2-(二丁基膦酰基)甲基-1,3-丙二醇(7a-2)的合成[化学式83]使2.50g合成例D5-2中得到的膦酸二丁酯化合物(6a-2)溶解于15.5ml甲醇中,进而加入442.5mg对甲苯磺酸一水合物,于20℃搅拌8小时。在减压下蒸馏除去反应液中的甲醇,将得到的残余物利用硅胶柱色谱法(氯仿∶甲醇=15∶1)进行纯化,以97%的收率得到了2.13g二元醇化合物(7a-2)。1H-NMR(500MHz,CDCl3)δ:0.94(t,J=7.5Hz,6H),1.41(tq,J=7.5,7.5Hz,4H),1.66(tt,J=8,8Hz,4H),1.90(dd,J=19,7Hz,2H),2.12(m,1H),3.76(m,4H),4.04(m,4H)合成例E2-3(工序E:R2=苄基)2-(二苄基膦酰基)甲基-1,3-丙二醇(7a-3)的合成[化学式84]使1.58g合成例D5-3中得到的膦酸二苄酯化合物(6a-3)溶解于8.1ml甲醇中,进而加入230.9mg对甲苯磺酸一水合物,于20℃搅拌3小时,然后,于0℃搅拌过夜。在减压下蒸馏除去反应液中的甲醇,将得到的残余物利用硅胶柱色谱法(氯仿∶甲醇=10∶1)进行纯化,以89%的收率得到了1.27g二元醇化合物(7a-3)。1H-NMR(500MHz,CDCl3)δ:1.92(dd,J=19,7Hz,2H),2.05(m,1H),3.70(m,4H),5.01(m,4H),7.34(m,10H)[工序(F)]合成例F1-1(工序F:碱=DBU,溶剂=DMF)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式85]使200.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于4ml的DMF中,进而加入45.3μl的DBU,于20℃搅拌3小时。向反应混合物中加入57.6mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿∶甲醇=9∶1)进行纯化,以88%的收率得到了147.1mg环状膦酸化合物(8a)。1H-NMR(500MHz,CDCl3)δ:1.73-2.07(m,3H),2.35(dd,J=5,5Hz,1H),2.68-2.88(m,1H),3.65-3.73(m,2H),3.79(dd,J=11,3.5Hz,3H),3.89-4.37(m,2H)合成例F1-2(工序F:碱=DBU,溶剂=DMAc)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式86]使500mg合成例E1-1中得到的二元醇化合物(7a)溶解于10.2ml二甲基乙酰胺中,进而加入113μl的DBU,于20℃搅拌3小时。用144mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的二甲基乙酰胺。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=5:1)进行纯化,以51%的收率得到了212mg环状膦酸化合物(8a)。合成例F1-3(工序F:碱=DBU,溶剂=乙腈(AN))(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式87]使500mg合成例E1-1中得到的二元醇化合物(7a)溶解于10.2ml乙腈中,进而加入113μl的DBU,于20℃搅拌3小时。用144mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的乙腈。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=10:1)进行纯化,以56%的收率得到了235mg环状膦酸化合物(8a)。合成例F1-4(工序F:碱=DBU,溶剂=丙酮)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式88]使500mg合成例E1-1中得到的二元醇化合物(7a)溶解于10.2ml丙酮中,进而加入113μl的DBU,于20℃搅拌3小时。用144mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的丙酮。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=10:1)进行纯化,以54%的收率得到了224mg环状膦酸化合物(8a)。合成例F1-4(工序F:碱=DBU,溶剂=甲醇)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式89]使500mg合成例E1-1中得到的二元醇化合物(7a)溶解于10.2ml甲醇中,进而加入113μl的DBU,于20℃搅拌3小时。用144mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的甲醇。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=10:1)进行纯化,以21%的收率得到了86.8mg环状膦酸化合物(8a)。合成例F1-5(工序F:碱=DBU,溶剂=异丙醇)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式90]使500mg合成例E1-1中得到的二元醇化合物(7a)溶解于10.2ml异丙醇中,进而加入113μl的DBU,于20℃搅拌3小时。用144mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的异丙醇。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=10:1)进行纯化,以18%的收率得到了75mg环状膦酸化合物(8a)。合成例F1-6(工序F:碱=DBU,溶剂=丁醇)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式91]使500mg合成例E1-1中得到的二元醇化合物(7a)溶解于10.2ml丁醇中,进而加入113μl的DBU,于20℃搅拌3小时。用144mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的丁醇。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=10:1)进行纯化,以29%的收率得到了121mg环状膦酸化合物(8a)。合成例F1-7(工序F:碱=DBU,溶剂=DMF/乙腈)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式92]使500mg合成例E1-1中得到的二元醇化合物(7a)溶解于5.1ml的DMF及5.1ml乙腈中,进而加入113μl的DBU,于20℃搅拌3小时。用144mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF及乙腈。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=10:1)进行纯化,以41%的收率得到了170mg环状膦酸化合物(8a)。合成例F2-1(工序F:碱=DBU(0.3eq.),溶剂=DMF)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式93]使500.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于10ml的DMF中,进而加入113μl的DBU,于20℃搅拌3小时。用144mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去DMF。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以73%的收率得到了304.7mg环状膦酸化合物(8a)。合成例F2-2(工序F:碱=DBN(0.3eq.),溶剂=DMF)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式94]使500.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于10ml的DMF中,进而加入90μl的DBN,于20℃搅拌4小时。用144mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以73%的收率得到了307.2mg环状膦酸化合物(8a)。合成例F2-3(工序F:碱=TEA(0.3eq.),溶剂=DMF)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式95]使500.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于10ml的DMF中,进而加入105μl的TEA,由20℃升温至加热回流,然后,搅拌2.5小时。用144mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以52%的收率得到了216.2mg环状膦酸化合物(8a)。合成例F2-4(工序F:碱=DIPEA(0.3eq.),溶剂=DMF)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式96]使500.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于10ml的DMF中,进而加入132μl的DIPEA,由20℃升温至加热回流,然后,搅拌2.5小时。用144mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以54%的收率得到了226.1mg环状膦酸化合物(8a)。合成例F2-5(工序F:碱=NaOMe(0.3eq.),溶剂=DMF)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式97]使500.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于10ml的DMF中,进而加入40.9mg甲醇钠,于20℃搅拌3小时。用144mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF。对于残余物,加入10ml氯仿,过滤白色的盐,将该白色固体用10ml氯仿洗涤,然后将滤液浓缩。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以62%的收率得到了258.1mg环状膦酸化合物(8a)。合成例F2-6(工序F:碱=NaOEt(0.3eq.),溶剂=DMF)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式98]将770.7mg合成例E1-1中得到的二元醇化合物(7a)溶解于15.4ml的DMF中,进而加入79.4mg乙醇钠,于20℃搅拌3小时。用221.9mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF。对于残余物,加入20ml氯仿,过滤白色的盐,将该白色固体用20ml氯仿洗涤,然后将滤液浓缩。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以54%的收率得到了349.0mg环状膦酸化合物(8a)。合成例F2-7(工序F:碱=t-BuOK(0.3eq.),溶剂=DMF)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式99]使500.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于10ml的DMF中,进而加入84.9mg叔丁醇钾,于20℃搅拌3小时。用144mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF。对于残余物,加入10ml氯仿,过滤白色的盐,将该白色固体用10ml氯仿洗涤,然后将滤液浓缩。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以46%的收率得到了193.8mg环状膦酸化合物(8a)。合成例F2-8(工序F:碱=t-BuONa(0.3eq.),溶剂=DMF)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式100]使500.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于10ml的DMF中,进而加入72.7mg叔丁醇钠,于20℃搅拌3小时。用144mg对甲苯磺酸一水合物使反应停止,过滤并除去析出的白色固体。将该白色固体用5ml的DMF洗涤,然后在减压下蒸馏除去滤液中的DMF。对于残余物,加入10ml氯仿,过滤白色的盐,用10ml氯仿洗涤,然后将滤液浓缩。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以48%的收率得到了200.6mg环状膦酸化合物(8a)。合成例F2-9(工序F:碱=CsCO3(0.3eq.),溶剂=DMF)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式101]使500.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于10ml的DMF中,进而加入246.6mg碳酸铯,于20℃搅拌3小时。用144mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF。对于残余物,加入10ml氯仿,过滤白色的盐,将该白色固体用10ml氯仿洗涤,然后将滤液浓缩。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以8%的收率得到了35.1mg环状膦酸化合物(8a)。合成例F2-10(工序F:碱=NaH(0.3eq.),溶剂=DMF)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式102]使500.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于10ml的DMF中,进而加入30.3mg氢化钠,于20℃搅拌3小时。用144mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF。对于残余物,加入10ml氯仿,过滤白色的盐,将该白色固体用10ml氯仿洗涤,然后将滤液浓缩。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以45%的收率得到了190.2mg环状膦酸化合物(8a)。合成例F3-1(工序F:淬灭剂=CSA)(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a)的合成[化学式103]使500.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于10ml的DMF中,进而加入113μl的DBU,于20℃搅拌3小时。用175.9mg樟脑磺酸(CSA)使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,定量地得到了554.2mg环状膦酸化合物(8a)。合成例F3-2(工序F:淬灭剂=甲酸)使500.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于10ml的DMF中,进而加入113μl的DBU,于20℃搅拌3小时。用30μl甲酸使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以73%的收率得到了305.6mg环状膦酸化合物(8a)。合成例F3-3(工序F:淬灭剂=乙酸)使500.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于10ml的DMF中,进而加入113μl的DBU,于20℃搅拌3小时。用43μl乙酸使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以69%的收率得到了289.6mg环状膦酸化合物(8a)。合成例F3-4(工序F:淬灭剂=丙酸)使500.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于10ml的DMF中,进而加入113μl的DBU,于20℃搅拌3小时。用57μl丙酸使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以72%的收率得到了303.0mg环状膦酸化合物(8a)。合成例F3-5(工序F:淬灭剂=丁酸)使500.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于10ml的DMF中,进而加入113μl的DBU,于20℃搅拌3小时。用69μl丁酸使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以72%的收率得到了303.3mg环状膦酸化合物(8a)。合成例F3-6(工序F:淬灭剂=三氟乙酸)使500.0mg合成例E1-1中得到的二元醇化合物(7a)溶解于10ml的DMF中,加入113μl的DBU,于20℃搅拌3小时。用58μl三氟乙酸使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以69%的收率得到了288.4mg环状膦酸化合物(8a)。合成例F4-1(工序F:碱=DBU,溶剂=乙腈(AN))(2-乙氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a-1)的合成[化学式104]使1.50g合成例E2-1中得到的二元醇化合物(7a-1)溶解于26.5ml的DMF中,进而加入297μl的DBU,于20℃搅拌3小时。进一步地,向该溶液中加入200μl的DBU,于20℃搅拌2小时。然后,用630.7mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化。使得到的级分再次溶解于25.1ml的DMF中,加入282μl的DBU,于20℃搅拌4小时。进一步地,向该溶液中补加282μl的DBU,于20℃搅拌3小时。接下来,补加282μl的DBU,于20℃搅拌3小时,用1.08g对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以69%的收率得到了781.6mg环状膦酸化合物(8a-1)。1H-NMR(500MHz,CDCl3)δ:1.35(t,J=7Hz,3H),1.75(m,1H),1.85(br,羟基),2.00(m,1H),2.15(br,羟基),2.78(m,1H),3.70(m,2H),3.89-4.36(m,4H)合成例F4-2(工序F:碱=DBU,溶剂=乙腈(AN))(2-乙氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a-1)的合成[化学式105]使1.50g合成例E2-1中得到的二元醇化合物(7a-1)溶解于26.5ml的DMF中,进而加入297μl的DBU,于20℃搅拌3小时。进一步地,向该溶液中加入200μl的DBU,于20℃搅拌2小时。然后,用630.7mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿∶甲醇=15∶1)进行纯化。使得到的级分再次溶解于25.1ml的DMF中,加入282μl的DBU,于20℃搅拌4小时。进一步地,向该溶液中补加282μl的DBU,于20℃搅拌3小时。接下来,补加282μl的DBU,于20℃搅拌3小时,用1.08g对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿∶甲醇=15∶1)进行纯化,以69%的收率得到了781.6mg环状膦酸化合物(8a-1)。合成例F4-3(工序F:R1=正丁基(n-Bu))(2-丁氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a-2)的合成[化学式106]使1.72g合成例E2-2中得到的二元醇化合物(7a-2)溶解于24.4ml的DMF中,进而加入911μl的DBU,于60℃搅拌5小时。然后,用1.16g对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿∶甲醇=10∶1)进行纯化,以81%的收率得到了1.03g环状膦酸化合物(8a-2)。1H-NMR(500MHz,CDCl3)δ:0.94(t,J=7.5Hz,6H),1.40(tq,J=7.5,7.5Hz,4H),1.64-1.79(m,3H),2.00(m,1H),2.36(br,羟基),2.69-2.89(m,1H),3.70(m,2H),3.89-4.36(m,2H),4.10(m,2H)合成例F4-4(工序F:R1=苄基)(2-苄氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇(8a-3)的合成[化学式107]使350.4mg合成例E2-3中得到的二元醇化合物(7a-3)溶解于4ml的DMF中,进而加入45μl的DBU,于20℃搅拌3小时。然后,用57.1mg对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去反应液中的DMF。将得到的残余物利用硅胶柱色谱法(氯仿∶甲醇=5∶1)进行纯化,以78%的收率得到了188.5mg环状膦酸化合物(8a-3)。1H-NMR(500MHz,CDCl3)δ:1.65-2.06(m,2H),2.66-2.80(m,1H),3.61-3.67(m,2H),3.88-4.36(m,2H),5.12(m,2H),7.33-7.41(m,5H)[工序(G)]合成例G1-1(工序G:溶剂=二氯甲烷)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式108]使300.0mg合成例F1-1中得到的环状膦酸化合物(8a)溶解于6.0ml二氯甲烷中,进而添加510.1mg油酸、及66.2mg的DMAP,冷却至0℃。然后,向该溶液中加入415.4mg的EDC,于室温搅拌2.5小时。用10ml的1N盐酸使反应停止,进行分液,然后用10ml二氯甲烷萃取,用5ml二氯甲烷再次萃取,用10ml的1%食盐水洗涤有机相。然后,在减压下蒸馏除去二氯甲烷,用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以84%的收率得到了649.8mg膦酸酯化合物(9a)。1H-NMR(500MHz,CDCl3):δ:0.88(t,J=6.5Hz,3H),1.27-1.30(m,20H,),1.60-1.76(m,3H)2.01-2.12(m,5H),2.32(t,J=7.5Hz,2H),2.83-2.97(m,1H),3.78-4.34(m,7H),5.31-5.38(m,2H)合成例G1-2(工序G:溶剂=甲苯)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式109]使300.0mg合成例F1-1中得到的环状膦酸化合物(8a)溶解于6.0ml甲苯中,进而添加510.1mg油酸、及66.2mg的DMAP,冷却至0℃。然后,向该溶液中加入415.4mg的EDC,于室温搅拌4小时。用10ml的1N盐酸使反应停止,添加2ml甲醇,进行分液,然后用10ml甲苯萃取3次,用10ml的1%食盐水洗涤有机相。然后,在减压下蒸馏除去甲苯,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以47%的收率得到了369.1mg膦酸酯化合物(9a)。合成例G1-3(工序G:溶剂=THF)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式110]使300.0mg合成例F1-1中得到的环状膦酸化合物(8a)溶解于6.0ml四氢呋喃中,进而添加510.1mg油酸、及66.2mg的DMAP,冷却至0℃。然后,向该溶液中加入415.4mg的EDC,于室温搅拌4小时。在减压下蒸馏除去四氢呋喃,分别加入10ml二氯甲烷、10ml的1N盐酸,然后进行分液,并用10ml二氯甲烷萃取2次,用10ml的1%食盐水洗涤有机相。然后,在减压下蒸馏除去二氯甲烷,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以66%的收率得到了509.6mg膦酸酯化合物(9a)。合成例G1-4(工序G:溶剂=DMF)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式111]使300.0mg合成例F1-1中得到的环状膦酸化合物(8a)溶解于6.0ml的N,N-二甲基甲酰胺中,进而添加510.1mg油酸、及66.2mg的DMAP,冷却至0℃。然后,向该溶液中加入415.4mg的EDC,于室温搅拌2.5小时。在减压下蒸馏除去N,N-二甲基甲酰胺,分别加入10ml二氯甲烷、10ml的1N盐酸,然后进行分液,用10ml二氯甲烷萃取2次,用10ml的1%食盐水洗涤有机相。然后,在减压下蒸馏除去二氯甲烷,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以78%的收率得到了603.4mg膦酸酯化合物(9a)。合成例G1-5(工序G:溶剂=乙酸乙酯)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式112]使300.0mg合成例F1-1中得到的环状膦酸化合物(8a)溶解于6.0ml乙酸乙酯中,进而添加510.1mg油酸、及66.2mg的DMAP,冷却至0℃。然后,向该溶液中加入415.4mg的EDC,于室温搅拌24小时。用10ml的1N盐酸进行淬灭后,进行分液,用10ml乙酸乙酯萃取2次,用10ml的1%食盐水洗涤有机相。然后,在减压下蒸馏除去乙酸乙酯,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以73%的收率得到了568.1mg膦酸酯化合物(9a)。合成例G2-1(工序G:缩合剂=DCC)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式113]使300.0mg合成例F1-1中得到的环状膦酸化合物(8a)溶解于6.0ml二氯甲烷中,进而添加510.1mg油酸。然后,将溶液冷却至0℃后,加入447.2mg的DCC,于室温搅拌23小时。通过过滤除去得到的白色固体,将白色固体用3ml二氯甲烷洗涤。然后,将滤液在减压下浓缩,并将残余物利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以3.5%的收率得到了27.1mg膦酸酯化合物(9a)。合成例G2-2(工序G:缩合剂=DCC,添加剂=DMAP)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式114]使300.0mg合成例F1-1中得到的环状膦酸化合物(8a)溶解于6.0ml二氯甲烷中,进而添加510.1mg油酸、及66.2mg的4-DMAP,冷却至0℃,然后加入447.2mg的DCC,于室温搅拌2小时。通过过滤除去得到的白色固体,用15ml二氯甲烷洗涤。然后,将滤液在减压下浓缩,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以59%的收率得到了458.0mg膦酸酯化合物(9a)。合成例G2-3(工序G:缩合剂=DIC)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式115]使300.0mg合成例F1-1中得到的环状膦酸化合物(8a)溶解于6.0ml二氯甲烷中,进而添加510.1mg油酸,冷却至0℃,然后加入339μl的DIC,于室温搅拌3天。通过过滤除去得到的白色固体,用5ml二氯甲烷洗涤。然后,将滤液在减压下浓缩,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以18%的收率得到了137.8mg膦酸酯化合物(9a)。合成例G2-4(工序G:缩合剂=DIC,添加剂=DMAP)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式116]使300.0mg合成例F1-1中得到的环状膦酸化合物(8a)溶解于6.0ml二氯甲烷中,进而添加510.1mg油酸、及66.2mg的4-DMAP,冷却至0℃。然后,向该溶液中加入339μl的DIC,于室温搅拌24小时。通过过滤除去得到的白色固体,并用5ml二氯甲烷洗涤。然后,将滤液在减压下浓缩,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以73%的收率得到了569.9mg膦酸酯化合物(9a)。合成例G3-1(工序G:使用了油酰氯的反应)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式117]使300.0mg合成例F1-1中得到的环状膦酸化合物(8a)溶解于6.0ml二氯甲烷中,进而添加376μl三乙胺,冷却至0℃,然后加入717μl油酰氯,于室温搅拌5.5小时。用10ml水进行淬灭,然后用10ml二氯甲烷萃取2次,将有机相用1%食盐水洗涤。减压下蒸馏除去二氯甲烷,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以85%的收率得到了658.4mg膦酸酯化合物(9a)。合成例G3-2(工序G:使用了制备的油酰氯的反应)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化118]使510.1mg油酸溶解于6.0ml甲苯中,进而添加7μl的N,N-二甲基甲酰胺及156μl亚硫酰氯,于40℃搅拌2小时。然后,在减压下蒸馏除去甲苯。进而加入5ml甲苯,并再次在减压下蒸馏除去甲苯,使残余物溶解于3.0ml二氯甲烷中。另一方面,使300.0mg环状膦酸化合物(8a)溶解于3.0ml二氯甲烷中,进而添加376μl三乙胺,进行冰冷却,然后滴加加入所制备的酰氯的二氯甲烷溶液3.0ml,于室温搅拌3小时。用10ml水进行淬灭,然后用10ml二氯甲烷萃取2次,将有机相用1%食盐水洗涤。在减压下蒸馏除去二氯甲烷,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以73%的收率得到了571.2mg膦酸酯化合物(9a)。合成例G3-3(工序G:使用了制备的油酰氯的反应)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式119]使510.1mg油酸溶解于6.0ml甲苯中,进而添加7μl的N,N-二甲基甲酰胺及190μl草酰氯,于40℃搅拌3.5小时,在减压下蒸馏除去甲苯。进而加入5ml甲苯,再次在减压下蒸馏除去甲苯,使残余物溶解于3.0ml二氯甲烷中。另一方面,使300.0mg环状膦酸化合物(8a)溶解于3.0ml二氯甲烷中,进而添加376μl三乙胺,进行冰冷却,然后滴加加入所制备的酰氯的二氯甲烷溶液3.0ml,于室温搅拌2小时。用10ml水进行淬灭,然后用10ml二氯甲烷萃取2次,将有机相用10ml水洗涤。在减压下蒸馏除去二氯甲烷,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以66%的收率得到了515.6mg膦酸酯化合物(9a)。合成例G4-1(工序G:使用了对甲苯磺酰氯的反应)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式120]使510.1mg油酸溶解于3.0ml二氯甲烷中,进而加入430μl的N-甲基咪唑(NMM)后,冷却至0℃后,于0℃搅拌1小时。然后,滴加加入3.0ml含有300.0mg环状膦酸化合物(8a)的二氯甲烷溶液,于0℃搅拌1.5小时。用10ml水进行淬灭后,用10ml二氯甲烷萃取2次,然后将有机相用1%食盐水洗涤。在减压下蒸馏除去二氯甲烷,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以78%的收率得到了603.8mg膦酸酯化合物(9a)。合成例G4-2(工序G:使用了向山试剂的反应)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式121]使421.9mg环状膦酸化合物(8a)及717.4mg油酸溶解于8.5ml二氯甲烷中,进而加入845μl三乙胺及778.7mg的2-氯-1-甲基吡啶鎓碘化物(CMPI),加热回流3小时。用10ml水进行淬灭,然后用10ml二氯甲烷萃取2次。在减压下蒸馏除去二氯甲烷,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以34%的收率得到了263.8mg膦酸酯化合物(9a)。合成例G4-3(工序G:使用了pyBop的反应)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式122]使200.0mg环状膦酸化合物(8a)及340.1mg油酸溶解于4.0ml二氯甲烷中,进而添加420μl二异丙基乙胺,进行冰冷却后,加入752mg的pyBop,并于室温搅拌6小时。接下来,用5ml的1N盐酸进行淬灭,然后依次用10ml二氯甲烷、5ml二氯甲烷萃取共2次。将有机相用10ml水洗涤,在减压下蒸馏除去二氯甲烷,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以99%的收率得到了513.8mg膦酸酯化合物(9a)。合成例G4-4(工序G:使用了HATU的反应)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式123]使300.0mg环状膦酸化合物(8a)及510.1mg油酸溶解于6.0ml二氯甲烷中,进而添加630μl二异丙基乙胺,进行冰冷却后,加入824.0mg的HATU,并于室温搅拌24小时。接下来,用10ml的1N盐酸进行淬灭,然后用10ml二氯甲烷萃取2次。将有机相用10ml水洗涤,在减压下蒸馏除去二氯甲烷,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以68%的收率得到了513.2mg膦酸酯化合物(9a)。合成例G4-5(工序G:使用了HBTU的反应)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式124]使300.0mg环状膦酸化合物(8a)及510.1mg油酸溶解于6.0ml二氯甲烷中,进而添加630μl二异丙基乙胺,进行冰冷却后,加入822.0mg的HBTU,于室温搅拌6.5小时。接着,用10ml的1N盐酸进行淬灭后,用10ml二氯甲烷萃取2次。将有机相用10ml水洗涤,在减压下蒸馏除去二氯甲烷,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而以79%的收率得到了612.0mg膦酸酯化合物(9a)。合成例G4-6(工序G:使用了COMU的反应)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式125]使300.0mg环状膦酸化合物(8a)及510.1mg油酸溶解于6.0ml二氯甲烷中,进而添加630μl二异丙基乙胺,进行冰冷却后,加入928.1mg的COMU,于室温搅拌24小时。接着,用10ml的1N盐酸进行淬灭后,用10ml二氯甲烷萃取2次。将有机相用10ml水洗涤,减压下蒸馏除去二氯甲烷,利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,从而定量地得到了778.2mg膦酸酯化合物(9a)。合成例G5(叠缩法)9-十八碳烯酸(9Z)-(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯(9a)的合成[化学式126](工序B)使30.0g化合物(3a)溶解于456ml的CH2Cl2中,加入42.7ml三乙胺,冷却至-20℃。向其中加入19.1ml的MsCl,于-20℃搅拌1小时。用250ml水使反应停止,分液后用150ml的CH2Cl2萃取水层,将有机层用200ml水洗涤。(工序C)将有机层在减压下浓缩,使得到的残余物溶解于684ml甲基乙基酮中,加入1.42ml三乙胺及46.14g碘化钠,使其在加热回流下反应2.5小时。将反应液冷却,然后在减压下蒸馏除去甲基乙基酮,加入300ml的CH2Cl2及300ml水,进行分液。将水层用150ml的CH2Cl2萃取2次,然后将有机层用2.5%硫代硫酸钠及300ml的0.5%碳酸氢钠水溶液洗涤。然后进行分液,接着用300ml水洗涤,将有机层在减压下浓缩。(工序D)使得到的残余物溶解于410ml的DMF中,进而加入133.73g碳酸铯及37.64ml亚磷酸二甲酯,于50℃使其反应3小时。在减压下蒸馏除去反应液中的DMF,加入300ml甲苯,过滤白色固体。(工序E)将白色固体用150ml甲苯洗涤,然后将滤液在减压下浓缩,使得到的残余物溶解于410ml甲醇中。进一步地,向该溶液中加入1.95g对甲苯磺酸一水合物,于20℃搅拌2小时。(工序F)将反应液在减压下浓缩,使得到的残余物溶解于821ml的DMF中。加入9.21ml的DBU,于20℃搅拌3小时。进一步地,向该溶液中加入11.71g对甲苯磺酸一水合物使反应停止,在减压下蒸馏除去DMF。(工序G)使得到的残余物溶解于684ml的CH2Cl2中,进而加入57.97g油酸、7.52g的DMAP及47.21g的EDC,于20℃使其反应12小时。接下来,加入300ml的1N盐酸,进行分液。用300ml的CH2Cl2将水层萃取2次,将有机层用300ml水洗涤,然后将有机层浓缩。将得到的残余物利用硅胶柱色谱法(仅用乙酸乙酯洗脱)进行纯化,以70%的收率(全部工序)得到了61.99g化合物(9a)。[工序(H)]实施例12ccPA的晶体的制造(良溶剂:水,不良溶剂:丙酮)使1.0g利用合成例G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应14小时。反应后,将反应液浓缩,接着于60℃将其溶解于5ml水中,冷却至20℃。然后,向该溶液中滴加20ml丙酮,熟化1小时。将晶体过滤,并用30ml丙酮洗涤,在减压下使其干燥,得到651.0mg纯度为98.874%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1728、1204、1176、1098、1012、774、744、721熔点:189℃实施例22ccPA的晶体的制造(良溶剂:甲醇,不良溶剂:丙酮)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应13小时。反应后,将反应液浓缩,接着于40℃将其溶解于2.5ml甲醇中,冷却至10℃。然后,向该溶液中滴加2.5ml丙酮。升温至20℃,然后滴加7.5ml丙酮,熟化1小时。然后,将晶体过滤,用30ml丙酮洗涤,在减压下使其干燥,得到907.6mg纯度为98.880%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1733、1209、1166、1097、1013、774、738、722熔点:188℃实施例32ccPA的晶体的制造(良溶剂:甲醇,不良溶剂:甲基乙基酮)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应13小时。反应后,将反应液浓缩,接着,于40℃将其溶解于2.5ml甲醇中,冷却至10℃。然后,向该溶液中滴加2.5ml甲基乙基酮。升温至20℃,然后滴加7.5ml甲基乙基酮,熟化1小时。然后,将晶体过滤,并用30ml甲基乙基酮洗涤,在减压下使其干燥,得到862.8mg纯度为98.944%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1727、1205、1175、1099、1024、773、742、721熔点:187℃实施例42ccPA的晶体的制造(良溶剂:甲醇,不良溶剂:甲基异丁基酮)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应12小时。反应后,将反应液浓缩,接着于40℃将其溶解于2.5ml甲醇中,冷却至10℃。然后,向该溶液中滴加2.5ml甲基异丁基酮。升温至20℃,然后滴加7.5ml甲基异丁基酮,熟化1小时。然后,将晶体过滤,并用30ml甲基异丁基酮洗涤,在减压下使其干燥,得到819.1mg纯度为99.300%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1735、1210、1165、1096、1012、776、738、722熔点:189℃实施例52ccPA的晶体的制造(良溶剂:甲醇,不良溶剂:甲基乙基酮:丙酮=1:1)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应13.5小时。反应后,将反应液浓缩,接着于40℃将其溶解于2.5ml甲醇中,冷却至10℃。然后,向该溶液中滴加2.5ml丙酮与甲基乙基酮的混合液。升温至20℃,然后滴加7.5ml丙酮与甲基乙基酮的混合液,熟化1小时。然后,将晶体过滤,并用30ml丙酮与甲基乙基酮的混合液洗涤,在减压下使其干燥,得到853.8mg纯度为98.880%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1733、1209、1166、1097、1013、775、738、722实施例62ccPA的晶体的制造(良溶剂:乙醇,析晶温度为10℃)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应13小时。反应后,将反应液浓缩,接着于60℃将其溶解于7.5ml乙醇中,冷却至10℃,熟化1小时。然后,将晶体过滤,并用30ml丙酮洗涤,在减压使其下干燥,得到846.6mg纯度为98.890%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1728、1207、1167、1097、1013、774、741、721熔点:189℃实施例72ccPA的晶体的制造(良溶剂:乙醇,析晶温度为20℃)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应16小时。反应后,将反应液浓缩,接着于65℃将其溶解于6ml乙醇中,冷却至20℃,熟化1小时。然后,将晶体过滤,并用30ml丙酮洗涤,在减压下使其干燥,得到783.3mg纯度为98.997%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1728、1211、1175、1096、1013、775、745、722熔点:190℃实施例82ccPA的晶体的制造(良溶剂:乙醇,不良溶剂:丙酮)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应13小时。反应后,将反应液浓缩,接着于60℃将其溶解于6ml乙醇中,冷却至20℃。向该溶液中滴加6ml丙酮后,熟化2小时。然后,将晶体过滤,并用30ml丙酮洗涤,在减压下使其干燥,得到786.4mg纯度为99.054%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1727、1212、1172、1096、1024、776、746、721熔点:189℃实施例92ccPA的晶体的制造(良溶剂:甲醇:乙醇=1:1,不良溶剂:丙酮)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应13.5小时。反应后,将反应液浓缩,接着于60℃将其溶解于2.5ml甲醇与乙醇的混合液中,冷却至10℃,滴加2.5ml丙酮后,升温至20℃,滴加7.5ml丙酮,熟化1小时,将晶体过滤,并用30ml丙酮洗涤,在减压下使其干燥,得到954.2mg纯度为98.812%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1734、1210、1165、1097、1012、775、738、722熔点:189℃实施例102ccPA的晶体的制造(良溶剂:1-丙醇,析晶温度为10℃)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应13小时。反应后,将反应液浓缩,接着于60℃将其溶解于7.5ml的1-丙醇中,冷却至10℃,熟化1小时。然后,将晶体过滤,并用70ml丙酮洗涤,在减压下使其干燥,得到645.0mg纯度为98.419%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1727、1207、1170、1098、1014、774、745、721熔点:190℃实施例112ccPA的晶体的制造(良溶剂:1-丙醇,析晶温度为20℃)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应14小时。反应后,将反应液浓缩,接着于50℃将其溶解于6.0ml的1-丙醇中,冷却至20℃,熟化1小时。然后,将晶体过滤,并用30ml丙酮洗涤,在减压下使其干燥,得到866.5mg纯度为98.750%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1727、1205、1174、1098、1023、773、743、721熔点:188℃实施例122ccPA的晶体的制造(良溶剂:1-丙醇,不良溶剂:丙酮)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应15.5小时。反应后,将反应液浓缩,接着于60℃将其溶解于6.0ml的1-丙醇中,冷却至20℃。向该溶液中滴加6.0ml丙酮,于20℃熟化1小时。然后,将晶体过滤,并用30ml丙酮洗涤,在减压下使其干燥,得到866.9mg纯度为98.902%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1727、1206、1175、1097、1023、774、744、721熔点:187℃实施例132ccPA的晶体的制造(良溶剂:异丙醇,析晶温度为20℃)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应15小时。反应后,将反应液浓缩,接着于65℃将其溶解于6.0ml的1-丙醇中,冷却至20℃,熟化1小时。然后,将晶体过滤,并用30ml丙酮洗涤,在减压下使其干燥,得到942.2mg纯度为98.588%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1727、1212、1175、1095、1023、776、746、722熔点:188℃实施例142ccPA的晶体的制造(良溶剂:异丙醇,不良溶剂:丙酮)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应14小时。反应后,将反应液浓缩,接着于60℃将其溶解于6.0ml异丙醇中,冷却至20℃。向该溶液中滴加6.0ml丙酮,于20℃熟化1小时。然后,将晶体过滤,并用30ml丙酮洗涤,在减压下使其干燥,得到915.8mg纯度为98.761%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2921、2851、1727、1212、1175、1095、1023、776、745、722熔点:187℃实施例152ccPA的晶体的制造(良溶剂:1-丁醇,不良溶剂:丙酮)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应12小时。反应后,将反应液浓缩,接着于65℃将其溶解于6.0ml的1-丁醇中,冷却至20℃。向该溶液中滴加6.0ml丙酮,于20℃熟化3.5小时。然后,将晶体过滤,并用30ml丙酮洗涤,在减压下使其干燥,得到598.9mg纯度为98.773%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1728、1206、1175、1097、1013、774、745、721熔点:189℃实施例162ccPA的晶体的制造(反应溶剂、析晶溶剂:丙酮)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml丙酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应48小时。冷却至室温,将晶体过滤,并用30ml丙酮洗涤,在减压下使其干燥,得到898.0mg纯度为86.921%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1727、1211、1175、1095、1023、776、746、722熔点:189℃实施例172ccPA的晶体的制造(反应溶剂、析晶溶剂:甲基乙基酮,不良溶剂:丙酮)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml丙酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应15小时。冷却至室温,并滴加11.6ml丙酮,将晶体过滤,并用30ml丙酮洗涤,在减压下使其干燥,得到950.7mg纯度为98.429%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1727、1211、1175、1096、1015、775、744、722熔点:189℃实施例182ccPA的晶体的制造(反应溶剂、析晶溶剂:甲基异丁基酮)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml丙酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应6小时。冷却至10℃,熟化1小时后,将晶体过滤,并用30ml丙酮洗涤,在减压下使其干燥,得到950.7mg纯度为20.777%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2921、2852、1731、1208、1174、1094、1011、778、745、722熔点:187℃实施例192ccPA的晶体的制造(良溶剂:甲醇,不良溶剂:乙酸乙酯)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应13小时。反应后,将反应液浓缩,接着于40℃将其溶解于2.5ml甲醇中,冷却至10℃,然后滴加2.5ml乙酸乙酯。升温至20℃,然后滴加7.5ml乙酸乙酯,熟化1小时。然后,将晶体过滤,并用30ml乙酸乙酯洗涤,在减压下使其干燥,得到719.8mg纯度为98.204%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1728、1208、1166、1097、1013、773、738、722熔点:190℃实施例202ccPA的晶体的制造(良溶剂:甲醇,不良溶剂:乙酸丁酯)使1.0g利用G5的制法而得到的环状膦酸酯(9a)溶解于11.6ml甲基乙基酮中,向其中加入522.3mg碘化钠,使其在加热回流下反应13小时。反应后,将反应液浓缩,接着于40℃将其溶解于2.5ml甲醇中,冷却至10℃,然后滴加2.5ml乙酸丁酯。升温至20℃,然后滴加7.5ml乙酸丁酯,熟化1小时,然后将晶体过滤,并用30ml乙酸丁酯洗涤,在减压下使其干燥,得到548.0mg纯度为98.350%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1735、1210、1165、1097、1012、776、738、722熔点:189℃实施例212ccPA的再纯化(良溶剂:甲醇,不良溶剂:甲基乙基酮)于40℃将3.00g实施例2中得到的2ccPA溶解于7.3ml甲醇中,将溶解液冷却至10℃,搅拌1小时,然后滴加7.3ml甲基乙基酮。然后,使其升温至20℃,再次滴加22ml甲基乙基酮,于20℃熟化1小时,然后,将晶体过滤,并用36ml甲基乙基酮洗涤,得到2.55g纯度为99.511%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1735、1210、1165、1096、1012、776、738、722熔点:189℃实施例222ccPA的再纯化(良溶剂:甲醇,不良溶剂:乙酸乙酯)于40℃将3.00g实施例2中得到的2ccPA溶解于7.3ml甲醇中,将溶解液冷却至10℃,并搅拌1小时,然后滴加7.3ml乙酸乙酯。然后,使其升温至20℃,再次滴加22ml乙酸乙酯,于20℃熟化1小时。然后,将晶体过滤,并用36ml乙酸乙酯洗涤,得到2.52g纯度为99.610%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1735、1210、1165、1097、1012、776、738、722熔点:187℃实施例232ccPA的再纯化(良溶剂:甲醇,不良溶剂:1-丙醇)于40℃将3.00g实施例2中得到的2ccPA溶解于7.3ml甲醇中,将溶解液冷却至10℃,搅拌1小时,然后滴加7.3ml的1-丙醇。然后,使其升温至20℃,再次滴加22ml的1-丙醇,于20℃熟化1小时。然后,将晶体过滤,并用36ml的1-丙醇洗涤,得到1.67g纯度为99.628%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1734、1210、1166、1097、1012、775、738、722熔点:187℃实施例242ccPA的再纯化(良溶剂:甲醇,不良溶剂:乙酸甲酯)于40℃将3.00g实施例2中得到的2ccPA溶解于7.3ml甲醇中,将溶解液冷却至10℃,并搅拌1小时,然后滴加7.3ml乙酸甲酯。然后,使其升温至20℃,再次滴加22ml乙酸甲酯,于20℃熟化1小时。然后,将晶体过滤,并用36ml乙酸甲酯洗涤,得到2.49g纯度为99.559%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1735、1210、1165、1097、1012、776、738、722熔点:189℃实施例252ccPA的再纯化(良溶剂:甲醇,不良溶剂:乙酸异丙酯)于40℃将3.00g实施例2中得到的2ccPA溶解于7.3ml甲醇中,将溶解液冷却至10℃,并搅拌1小时,然后滴加7.3ml乙酸异丙酯。然后,使其升温至20℃,再次滴加22ml乙酸异丙酯,并于20℃熟化1小时。然后,将晶体过滤,并用36ml乙酸异丙酯洗涤,得到2.37g纯度为99.549%的2ccPA。测定得到的2ccPA白色晶体的X射线粉末衍射光谱,其结果如下。该X射线粉末衍射光谱是利用通过单色仪后的的铜射线而得到的X射线粉末衍射光谱。IR光谱(cm-1):2920、2851、1735、1210、1165、1097、1012、776、738、722熔点:187℃实施例26(2ccPA的合成:反应溶剂为丙酮)[化学式127]使200mg环状膦酸酯化合物(9a)溶解于2.3ml丙酮中,向其中加入104.5mg碘化钠,使其在加热回流下反应23小时。冷却至20℃,然后将生成的白色固体过滤。将晶体用丙酮洗涤后,得到174.2mg(熔点为189.6℃)2ccPA(1)的白色晶体。1H-NMR(500MHz,CDCl3)δ:0.79(t,J=6.5Hz),1.19-1.23(m,20H),1.36(m,1H),1.51(br,2H),1.79(m,1H),1.93(br,4H),2.26(t,J=7.5Hz,2H),2.72(m,1H),3.65-4.10(m,4H),5.20-5.28(m,2H)[按照文献方案的合成研究]比较例1:膦酸二甲酯的合成、阿尔布佐夫反应[化学式128]使15.00g碘化合物(5a)溶解于105ml亚磷酸三甲酯中,并加热回流14小时。然后,再次加入210ml亚磷酸三甲酯,并加热回流6小时。浓缩后,将得到的残余物利用硅胶柱色谱法(氯仿∶甲醇=15∶1)进行纯化,以60%的收率得到了8.34g膦酸二甲酯化合物(6a),但混入了结构未知的副产物。比较例2:(2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲醇的合成、环化反应[化学式129]使8.34g比较例1中得到的膦酸二甲酯化合物(6a)溶解于417ml甲苯、及14.1ml甲醇中,加入1.53g对甲苯磺酸一水合物,加热回流3小时。在减压下蒸馏除去甲苯及甲醇,并利用硅胶柱色谱法(氯仿:甲醇=15:1)进行纯化,以42%的收率得到了2.44g环状膦酸化合物(8a)。比较例3:(9-十八碳烯酸-2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯的合成、缩合反应[化学式130]使2.42g比较例2中得到的环状膦酸化合物(8a)、及4.11g油酸、534.0mg的4-二甲基氨基吡啶溶解于48.6ml二氯甲烷中。将其进行冰冷却,然后,加入3.35g的EDC、1.23g油酸、2.23g的EDC、及808ml二氯甲烷,并于室温搅拌24小时。用571ml甲醇稀释,然后加入300ml水。分液后,将水相依次用300ml乙酸乙酯、100ml乙酸乙酯萃取共2次,将有机相用硫酸镁干燥,然后在减压下蒸馏除去乙酸乙酯、甲醇,将得到的残余物利用硅胶柱色谱法(仅乙酸乙酯)进行纯化,以42%的收率得到了2.65g环状膦酸酯化合物(9a)。比较例4:(9-十八碳烯酸-2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯质子型的合成、脱甲基化反应[化学式131]使2.51g比较例3中合成的环状膦酸酯化合物(9a)溶解于303ml二氯甲烷中,冷却至-15℃,然后加入2.31ml三甲基溴硅烷,于-15℃搅拌4.5小时。将反应液注入200ml冰水中,用750ml乙醚萃取并分液,然后,再次用200ml乙醚萃取2次。将有机相用硫酸钠干燥,在减压下蒸馏除去溶剂,然后,利用硅胶柱色谱法(氯仿:甲醇=5:1)进行纯化,以15%的收率得到了367.6mg化合物(10a:2ccPA的质子型)。比较例5:(9-十八碳烯酸-2-甲氧基-2-氧代-2λ5-[1,2]氧磷杂环戊烷-4-基)甲酯钠盐的合成[化学式132]使418.6mg比较例4中得到的化合物(10a)溶解于30ml乙醚中,加入20ml的0.05M的氢氧化钠水溶液并搅拌,然后进行分液。将水相冷冻干燥,由此以57%的收率得到了250.8mg的2ccPA(1)(纯度为67.934%)。试验例1:35℃稳定性试验将实施例3中得到的2ccPA的晶体(本发明)、比较例5中得到的2ccPA(文献方案)及无定形的2ccPA分别于35℃保存1个月,实施稳定性试验。将结果示于下述表1。上述无定形的2ccPA使用了将300mg实施例3中得到的2ccPA溶解于5ml水中并进行冷冻干燥而得到的物质。稳定性试验为:分别称量约15mg本发明的2ccPA的晶体、文献方案所得的2ccPA及无定形的2ccPA,用5ml的乙腈/水(1/1)进行稀释,利用岛津制作所LC-2010CHT,每隔一周对5μl这些稀释液进行分析。[表1]天数071421284256文献方案的2ccPA67.934%66.665%67.517%62.77%59.841%本发明的2ccPA的晶体99.561%99.605%99.576%99.631%99.626%99.593%99.493%无定形的2ccPA99.599%83.363%45.02%34.738%27.077%如上述表1的结果所示,按照比较例1~5的文献方案制得的2ccPA的纯度差、不稳定,无定形的2ccPA虽然纯度好,但分解逐周进展,不稳定。另一方面,利用本发明的制造方法得到的2ccPA的晶体为高纯度且稳定的晶体。即使在56天后仍维持99.493%的纯度,与利用现有的制造方法得到的2ccPA相比,具有优异的保存稳定性。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1