作为抗癌药物的功能化的和取代的吲哚的利记博彩app
本发明广泛涉及作为增殖性疾病如癌症,和一系列退行性疾病如骨关节炎、动脉粥样硬化、心脏病和炎症性肠病治疗的药物。本发明尤其涉及包含芳基和/或烷基取代的吲哚类化合物的药物。本发明进一步涉及治疗或预防疾病或病症如增殖性疾病(尤其是癌症)的方法。本发明还涉及制备所述化合物的工艺方法。
发明背景
本发明说明书参考任何现有技术并非承认或建议,所述现有技术在任何管辖地均组成常见的一般知识的一部分,或对于本领域技术人员而言,所述现有技术和/或结合其他现有技术,可以合理地应该被理解、被认为是相关的。
癌症已杀死成千上万的人,并在美国成为第二大死因。如今,在治疗或预防多种多样的癌症中已取得重大突破。例如,乳腺癌患者已从早期筛查项目以及各种外科手术技术中获益。然而,这些通常证明使人在身体上和情感上衰弱。并且,经历手术和后续化疗的患者,在他们所患疾病上通常会经历复发。
一种特定攻击癌细胞的潜在的新方法是通过破坏癌细胞的细胞骨架系统,其主要包含在肌动蛋白中。所述肌动蛋白细胞骨架密切地参与细胞分裂和细胞迁移。然而,肌动蛋白作为肿瘤细胞的细胞骨架和肌肉肌小节的肌动蛋白微丝,起着无处不在的作用。不同的角色但相似的结构使得肌动蛋白成为药物研发中较难的靶标,由于其多余的脱靶副反应。
发明摘要
本发明寻求解决上述问题中的一个或多个,和/或提供治疗改善(如癌症治疗),并在其中一个实施例提供抗原肌球蛋白的化合物。
第一方面,本发明提供通式(I)所示化合物,或其药学上可接受的药物或前药,
其中:
R1和R2独立地为H或C1-C6烷基;
R3是N(R7)2或3至7个原子组成的碳环,其中1至3个环碳原子可任选地被S、N、O、NH或NR7替换,和其中所述环可任选地被R7取代;
R4和R5独立地为或5或6个原子组成的碳环,其中1至3个环碳原子可任选地被S、N、O、NH或NR8替换,和其中所述环可任选地被R8取代;
R6是C1-C6烷基基团、C2-C6烯烃基团、或具有5至10个环碳原子的单环或双环的碳环,其中1或2个环碳原子可任选地被S、O、N、NH或NR7替换,和其中所述环可任选地被R8取代,或R6是
X1不存在,或X1是具有1至10个碳原子的烷基基团,或具有2至10个碳原子的烯基基团;
X2、X3和X4独立地不存在,或独立地选自由以下基团组成的组:S、O、NH、NHR7、C(O)、C(O)NH、具有1至10个碳原子的烷基基团、具有2至10个碳原子的烯烃基团、CH(R7)CHC(R7)C(O)、(CH2)0-5C(R7)C(R7)(CH2)0-5、和5或6个原子组成的碳环,其中1至3个环碳原子可任选地被S、N、O、NH或NR7替换;
X5是O、NH、NR7或S;
R7是H、C1-C6烷基、(CH2)1-5OMe、CF3、CN或OCF3;和
R8是H、OH、烷基(如C1-C6烷基)、烯基(如C2-C6烯基)、卤基、烷氧基、氨基、烷基氨基、二烷基氨基、或与R4、R5或R6上的两个相邻碳原子并合的二氧戊环。
X1可以是具有1至10个碳原子(如具有1至5个碳原子)的烷基基团。
R3可以是N(R7)2或4、5、6或7个原子组成的碳环(如环烷基),其中1至3个环碳原子可任选地被S、N、O、NH或NR7替换,和其中所述环可任选地被R7取代。
R1和R2可独立地为C1-C6烷基(如R1可以是,譬如,CH3或CH2CH3,和R2可以是,譬如,CH3或CH2CH3)。
X2、X3和X4可独立地选自由以下基团组成的组:S、O、NH、NHR7、C(O)、C(O)NH、具有1至10个碳原子(如1至5个碳原子)的烷基基团、CH(R7)CHC(R7)C(O)、(CH2)0-5C(R7)C(R7)(CH2)0-5、和5个原子组成的碳环(如芳基),其中1至3个环碳原子(如1或2个环碳原子)可任选地被S、N、O、NH或NR7(如N和/或O)替换。
R4和R5可独立地为5或6个原子组成的芳基或环烷基基团,其中1至3个环碳原子可任选地被S、N、O、NH或NR8替换,和其中所述环可任选地被R8取代。
R6可以是C1-C6烷基基团(如CH3或CH2CH3)或具有6至10个环碳原子的单环或双环的芳基基团,其中1或2个环碳原子可任选地被S、O、N、NH或NR7替换,和其中所述环可任选地被R8取代。R6可以是:
在其中一个实施例,所述式(I)的化合物,或其药学上可接受的药物或前药,是:
其中:
R1和R2可以均为CH3或CH2CH3。
X1可以是具有1至5个碳原子的烷基基团(如CH2、(CH2)2或(CH2)3)。
R3可以是4、5、6、或7个原子组成的环烷基基团,其中1至3个环碳原子可任选地被S、N、O、NH或NR7替换,和其中所述环可任选地被R7取代,譬如:
R3可以是6个原子组成的环烷基基团,其中1至3个环碳原子可任选地被S、N、O、NH或NR7替换,和其中所述环可任选地被R7取代,譬如:
X5可以是NH或NR7。R7可以是C1-C6烷基(如CH3或CH2CH3)。
X2可以是具有1至10个碳原子的烷基基团、O或NH。X2可以是(CH2)1-5(如CH2、(CH2)2或(CH2)3)。
R4可以是5或6个原子组成的芳基基团,其中1至3个环碳原子可任选地被S、N、O、NH或NR8替换,和其中所述环可任选地被R8取代,譬如:
R8可以是H。
X3可以是C(O)。
R5可以是5或6个原子组成的环烷基基团,其中1至3个环碳原子可任选地被S、N、O、NH或NR8替换,和其中所述环可任选地被R8取代,譬如:
X4可以是具有1至5个碳原子的烷基基团(如CH2、(CH2)2或(CH2)3)。
R6可以是具有9或10个环碳原子的双环芳基基团,其中1或2个环碳原子可任选地被S、O、N、NH或NR7替换,和其中所述环可任选地被R8取代。R6可以选自:
R8可以是选自H、烷氧基、卤基、和与R6上的两个相邻碳原子并合的二氧戊环。R8可以是烷氧基(如OCH3或OCH2CH3)。R8可以是卤基(如氟)。
尤其,本发明第一方面的所述化合物体现在以下结构:
在其中一个实施例,所述化合物是:
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮
(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-苯乙基哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮
(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-苯乙基哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮
(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-苯乙基哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮
(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-苯乙基哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮
(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-苯乙基哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮
(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-苯乙基哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮
3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(6-甲氧基萘-2-基)乙基)哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-1-基)乙基)哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(7-甲氧基萘-1-基)乙基)哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(喹啉-6-基)乙基)哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(6-甲氧基萘-2-基)乙基)哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-1-基)乙基)哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(7-甲氧基萘-1-基)乙基)哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(喹啉-6-基)乙基)哌嗪-1-基)甲酮
(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(喹啉-7-基)乙基)哌嗪-1-基)甲酮
(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(喹啉-7-基)乙基)哌嗪-1-基)甲酮。
第二方面,本发明涉及药物组合物,其包含式(I)所示化合物以及药学上可接受的载体、稀释剂或赋形剂。
本发明所述化合物和药物组合物可适于增殖性疾病的治疗或预防。相应地,另一方面,本发明涉及治疗或预防患者增殖性疾病的方法,所述方法包含给予所述患者有效量的根据本发明第一方面所述式(I)化合物或根据本发明第二方面所述药物组合物。
在进一步方面,本发明涉及根据本发明第一方面所述式(I)化合物或根据本发明第二方面所述药物组合物在制备用于治疗或预防增殖性疾病的药物中的用途。
在进一步方面,本发明涉及根据本发明第一方面所述式(I)化合物或根据本发明第二方面所述药物组合物用于患者增殖性疾病的治疗或预防的用途。
在进一步方面,本发明涉及根据本发明第一方面所述式(I)化合物或根据本发明第二方面所述药物组合物用于患者增殖性疾病的治疗或预防。
在一个或多个优选实施例,所述增殖性疾病是癌症,优选固体肿瘤。在各种优选实施例,所述癌症是选自由乳腺癌、肺癌、前列腺癌、卵巢癌、子宫癌、脑癌、皮肤癌、结肠癌和膀胱癌组成的组。
本领域技术人员应理解在本发明上下文中,“有效量”是指对被治疗患者给予足够量,从而产生所需的治疗或药理效应。
在进一步方面,本发明涉及完全或部分预防患者固体肿瘤复发的方法,所述方法包含给予患者有效量的根据本发明第一方面所述的式(I)化合物或根据本发明第二方面所述药物组合物。
另一方面,本发明涉及根据本发明第一方面所述的化合物或根据本发明第二方面所述药物组合物在制备用于完全或部分预防固体肿瘤复发的药物中的用途。
在进一步方面,本发明涉及根据本发明第一方面所述的式(I)化合物或根据本发明第二方面所述药物组合物用于完全或部分预防患者固体肿瘤复发的用途。
在进一步方面,本发明涉及根据本发明第一方面所述的式(I)化合物或根据本发明第二方面所述药物组合物用于完全或部分预防患者固体肿瘤的复发。
本发明所述化合物或药物组合物可适于炎症性疾病或病症的治疗或预防。相应地,另一方面,本发明涉及治疗患者炎症性疾病或病症的方法,所述方法包含给予所述患者有效量的根据本发明第一方面所述式(I)化合物或根据本发明第二方面所述药物组合物。
在进一步方面,本发明涉及根据本发明第一方面所述式(I)化合物或根据本发明第二方面所述药物组合物在制备用于治疗炎症性疾病或病症的药物中的用途。
在进一步方面,本发明涉及根据本发明第一方面所述式(I)化合物或根据本发明第二方面所述药物组合物用于患者炎症性疾病或病症的治疗的用途。
在进一步方面,本发明涉及根据本发明第一方面所述式(I)化合物或根据本发明第二方面所述药物组合物用于患者炎症性疾病或病症的治疗。
所述炎症性疾病或病症可以是骨关节炎、炎症性肠病(如溃疡性结肠炎和克罗恩氏病)、溃疡性直肠炎、远端结肠炎、自身免疫性疾病(如SLE、类风湿性关节炎、肾小球性肾炎)、哮喘或涉及肺部炎症的疾病、或心血管疾病(如动脉粥样硬化、高血压、和血脂异常)。
所述式(I)的化合物可以用于单独治疗或联合一个或多个其他药物(如化学治疗剂或抗炎剂)进行治疗,例如作为联合治疗的一部分。
另一方面,本发明涉及制备式(I)所示化合物的工艺方法,包括以下步骤:
另一方面,本发明涉及制备式(I)所示化合物的工艺方法,包括以下步骤:
另一方面,本发明涉及制备式(I)所示化合物的工艺方法,包括以下步骤:
本发明的进一步方面和上述段落中所描述方面的进一步实施例,将通过下述描述、给出的例子和参照附图从而变得显而易见。
附图简要说明
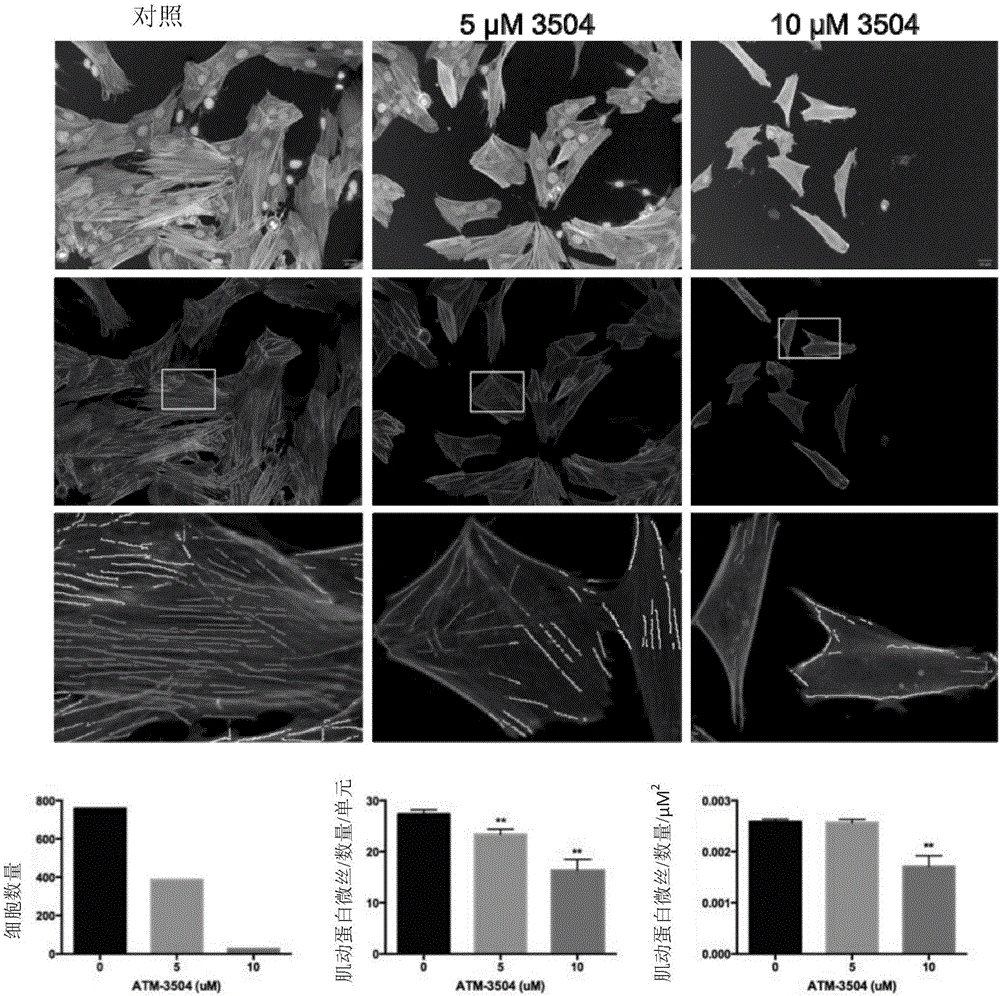
图1:肌动蛋白微丝在通过化合物(A)3504、(B)3507和(C)3516处理的SK-N-SH神经母细胞瘤细胞中的成像和定量。细胞被488-Atto-Phallodin和DAPI染色从而分别显现肌动蛋白微丝束和细胞核。显示在顶部板是来自对照(单独媒介物)的代表性的灰度免疫荧光图像,5μM和10μM处理的细胞。中间板(与放大插图显示在底部板)显示具有定量线性特征的细胞图像的叠加。彩色线表示检测到的肌动蛋白微丝。同时还显示的是细胞数量、单丝数量/单元和单丝数量/单元面积(μM2)的定量。统计分析是使用ANNOVA-多重比较的一种方法来执行完成,其中每个药物治疗组均与所述对照进行比较。****p<0.0001,****p<0.001,***p<0.01,**p<0.1。
图2:肌动蛋白微丝在通过化合物(A)3504、(B)3507和(C)3516处理的SK-N-SH神经母细胞瘤细胞中的成像和定量。细胞被γ9d(羊多克隆,1:100)染色,随后被488-共轭的第二抗体(1:1000)和DAPI染色,从而分别显现包含丝束和细胞核的Tpm3.1。显示在顶部板是来自对照(单独媒介物)的代表性的灰度免疫荧光图像,5μM和10μM处理的细胞。中间板(与放大插图显示在底部板)显示具有定量线性特征的细胞图像的叠加。彩色线表示检测到的肌动蛋白微丝。同时还显示的是细胞数量、单丝数量/单元和单丝数量/单元面积(μM2)的定量。统计分析是使用ANNOVA-多重比较方法的一种来执行完成,其中每个药物治疗组均与所述对照进行比较。****p<0.0001,****p<0.001,***p<0.01,**p<0.1。
图3:化合物3507对Tpm3.1-调节的肌动蛋白微丝解聚作用动力学的影响。6μM肌动蛋白微丝(35%芘标记),存在或不存在Tpm3.1的饱和量(10μM)情况下,稀释12倍成F-肌动蛋白缓冲液(100mM NaCl,10mM Tris-HCl pH 7.0,2mM MgCl2,1mM EGTA,0.2mM CaCl2,0.2mM ATP,0.5mM DTT,0.01%(v/v)NaN3)的(A和B)解聚作用时间进程。F-肌动蛋白和Tpm3.1的终浓度分别为0.5μM和0.83μM。Tpm3.1在与F-肌动蛋白混合之前,用50μM化合物3507或1%(v/v)DMSO预孵育。解聚作用数据归一化至初始荧光值。在化合物4015或4093存在的情况下,对于单独F-肌动蛋白或Tpm3.1/F-肌动蛋白的解聚作用的(C和D)初始速率(V0)。解聚作用的初始速率从开始的3600s进行测定,适于线性回归模型。数据代表平均值±SEM,平均从n>6重复。****p<0.0001。
图4:在神经母细胞瘤的侧翼异种移植模型(CHLA-20)中,将化合物3507/30%w/v Dexolve-7每天以150mg/kg进行IP给药,并给药18天。肿瘤体积每2-3天进行测定。
图5:在黑色素瘤的侧翼异种移植模型(A375)中,将化合物3507/30%w/v Dexolve-7以2x/周并以60mg/kg进行IV给药,并给药14天。肿瘤体积每2-3天进行测定。**p<0.01。
本发明实例的详细说明
本发明是基于惊人地发现通式(I)所示化合物能有效抑制原肌球蛋白,从而在治疗增殖性疾病,尤其是癌症方面得到了意想不到的改善结果。所述肌动蛋白细胞骨架的研究涉及大量辅助控制和调节蛋白。与癌症细胞的细胞骨架有关的肌动调节蛋白的识别和特定靶向,提供了研发特定癌症药物但没有不必要副反应的机会。
肌动蛋白微丝是通过球状肌动蛋白蛋白单体的聚合作用来构建的。所述肌动蛋白单体通过一端承载正电荷,而另一端承载负电荷来进行两极化。所述肌动蛋白微丝因此具有所有在同一方向对齐的所述肌动蛋白的蛋白。所述这些微丝具有与其相关的二级螺旋蛋白,原肌球蛋白。所述原肌球蛋白在调节肌动蛋白微丝功能中扮演着不可或缺的角色。在结构上,所述肌动蛋白微丝是由聚合的肌动蛋白单体和原肌球蛋白二聚体置于所述肌动蛋白微丝的α螺旋槽,从而形成均聚物。已有超过40种哺乳动物原肌球蛋白亚型,每一种均调节特定的肌动蛋白微丝。所述特定的原肌球蛋白亚型调节癌症细胞的细胞骨架,破坏这种相互作用,从而提供特异性治疗癌症细胞的基础。
I.定义
以下是一些用于本领域术语的定义,可以帮助理解本发明。这些术语均作为一般定义,且这些术语单独不应该限制本发明的范围,仅提出用于更好地理解本发明下述内容。
除非上下文另有要求或具体声明为相反内容,否则本发明所列举整数、步骤、或要素均作为单独的整数、步骤或要素,显然包括单数和复数形式的整数、步骤或要素。
本领域技术人员应领会本发明易受变化和改变而非那些具体描述的影响。应理解本发明包括所有这些变化和改变。本发明也包括所有本发明说明书所提及或表明的步骤、特征、组合物和化合物,单独地或集体地,任何及任何两个或多个所述步骤、特征、组合物和化合物的所有组合。
本发明使用的术语“包含”和“包括”为开放式和非限制性表达,除非另有说明。
贯穿本发明的术语“任选取代的”表示所述基团可能或可能不进一步被一个或多个非氢原子取代基团取代或并合(以便形成多环体系)。对于一个特定的官能团,合适的化学上可行的任选取代基对于本领域技术人员而言是显而易见的。典型的任选取代基包括C1-C4烷基、C2-C4烯基、OH、卤素、O(C1-C4烷基)、NRaRb其中Ra和Rb各自独立地选自H、C1-C3烷基、CONH2、SH、S(C1-C3烷基)、-CH2-O(C1-3烷基)、C6-10芳基、-CH2-苯基、羟基-(C1-3烷基)、和卤基-(C1-3烷基)。目前优选的任选取代基包括C1-3烷基、C1-3烷氧基、-CH2-(C1-3)烷氧基、C6-10芳基、-CH2-苯基、卤素、OH、羟基-(C1-3)烷基、和卤基-(C1-3)烷基,如CF3、CH2CF3。
“酰基”表示烷基-CO-基团,其中烷基基团具有如本发明所述的定义。酰基实例包括乙酰基和苯甲酰基。所述烷基基团可以是C1-C6烷基、C1-C4烷基、或C1-C3烷基基团。所述基团可以是末端基团或桥接基团。
“烷基”作为一个基团或基团的一部分是指具有1-12个碳原子、或1-10个碳原子、或1-6个碳原子、或1-4个碳原子、或1-3个碳原子的直链或支链脂肪族烃基团。因此,术语烷基的实例包括但不限于甲基、乙基、1-丙基、异丙基、1-丁基、2-丁基、异丁基、叔丁基、戊基、1,2-二甲基丙基、1,1-二甲基丙基、戊基、异戊基、己基、4-甲基戊基、1-甲基戊基、2-甲基戊基、3-甲基戊基、2,2-二甲基丁基、3,3-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基、1,2,2-三甲基丙基、1,1,2-三甲基丙基、2-乙基戊基、3-乙基戊基、庚基、1-甲基己基、2,2-二甲基戊基、3,3-二甲基戊基、4,4-二甲基戊基、1,2-二甲基戊基、1,3-二甲基戊基、1,4-二甲基戊基、1,2,3-三甲基丁基、1,1,2-三甲基丁基、1,1,3-三甲基丁基、5-甲基庚基、1-甲基庚基、辛基、壬基、癸基等等。所述基团可以是末端基团或桥接基团。
“烯基”作为一个基团或基团的一部分是指脂肪族烃基团,其包含至少一个碳-碳双键,且其可以是直链或支链的,例如在正常链中,一个基团具有2-12个碳原子、或2-6个碳原子、或2-4个碳原子。在正常链中,所述基团可以包含多个双键,且每个双键的定位各自独立地为顺式或反式,E或Z。烯基基团的实例包括但不限于次乙基、乙烯基、烯丙基、1-甲基乙烯基、1-丙烯基、2-丙烯基、2-甲基-1-丙烯基、2-甲基-1-丙烯基、1-丁烯基、2-丁烯基、3-丁烯基、1,3-丁二烯基、1-戊烯基、2-戊烯基、3-戊烯基、4-戊烯基、1,3-戊二烯基、2,4-戊二烯基、1,4-戊二烯基、3-甲基-2-丁烯基、1-己烯基、2-己烯基、3-己烯基、1,3-己二烯基、1,4-己二烯基、2-甲基戊烯基、1-庚烯基、2-庚烯基、3-庚烯基、1-辛烯基、1-壬烯基、1-癸烯基等等。所述基团可以是末端基团或桥接基团。
“烯基氧基”是指-O-烯基基团,其中烯基具有如本发明所述的定义。优选的烯基氧基基团是C2-C12烯基氧基基团。所述基团可以是末端基团或桥接基团。
术语“烷基氧基”和“烷氧基”为同义词,是指-O-烷基基团,其中烷基具有本发明所述的定义。目前优选的烷氧基基团是C1-6烷氧基或C1-4烷氧基或C1-3烷氧基。其实例包括但不限于甲氧基、乙氧基、n-丙氧基、异丙氧基、2-丁氧基、叔丁氧基等等。所述基团可以是末端基团或桥接基团。
“烷基氨基”或“烷氨基”包括单烷基氨基和二烷基氨基,除非另有说明。“单烷基氨基”是指–NH-烷基基团,其中烷基具有本发明上述的定义。“二烷基氨基”是指–N(烷基)2基团,其中每个烷基可以相同或不同,并且每个烷基具有本发明所述的定义。所述烷基基团可以是C1-C6烷基基团。所述基团可以是末端基团或桥接基团。
“炔基”作为一个基团或基团的一部分是指脂肪族烃基团,其包含碳-碳三键,且其在正常链中,可以是直链或支链的可具有2-12个碳原子、或2-6个碳原子、或2-4个碳原子。结构实例包括但不限于乙炔基和丙炔基。所述基团可以是末端基团或桥接基团。
“炔基氧基”是指-O-炔基基团,其中炔基具有本发明所述的定义。目前优选的炔基氧基基团是C2-C6炔基氧基基团、C2-C4炔基氧基。所述基团可以是末端基团或桥接基团。
“芳基”作为一个基团或基团的一部分是指(i)任选取代的单环、或并合多环、芳香的碳环(环结构上的环原子均为碳原子),其中每个环具有5-18个原子。目前优选的芳基基团的每个环具有6-14个原子,或更加优选每个环具有6-10个原子。芳基基团的实例包括苯基、萘基、菲基等等;(ii)任选取代的部分饱和的双环芳香碳环部分,其中苯基与C5-7环烷基或C5-7环烯基基团并合在一起形成环结构,例如四氢萘基、茚基或茚满基。所述基团可以是末端基团或桥接基团。
“环烯基”是指非芳香性单环或多环环系包含至少一个碳-碳双键,且每个环可以有5-10个碳原子。单环环烯基的实例包括环戊烯基、环己烯基或环庚烯基。所述环烯基基团可以被一个或多个取代基团取代。所述基团可以是末端基团或桥接基团。
“环烷基”是指饱和或部分饱和的单环或并合多环或螺多环的碳环,其中每个环可以包含3至9个碳原子,譬如环丙基、环丁基、环戊基、环己基等等,除非另有说明。所述环烷基包括单环体系如环丙基和环己基,双环体系如十氢萘,和多环体系如金刚烷。所述基团可以是末端基团或桥接基团。
本发明使用的术语“碳环”是指基于碳原子的环体系,其旨在包括如本发明所定义的芳基、环烯基、环烷基、和杂芳基基团。
术语“卤素”或“卤基”为同义词,均指氟、氯、溴或碘。
“杂芳基”无论单独使用或作为基团的一部分是指包含芳香环(例如5-或6-个原子组成的芳香环)的基团,并在所述芳香环上具有一个或多个杂原子作为环原子,且余下环原子为碳原子。合适的杂原子包括氮、氧或硫原子。杂芳基的实例包括噻吩、苯并噻吩、苯并呋喃、苯并咪唑、苯并噁唑、苯并噻唑、苯并异噻唑、萘并[2,3-b]噻吩、呋喃、异吲嗪、二苯并呋喃哌嗪(xantholene)、氧硫杂蒽酮(phenoxatine)、吡咯、咪唑、吡唑、吡啶、吡嗪、嘧啶、哒嗪、吲哚、异吲哚、1H-吲唑、嘌呤、喹啉、异喹啉、酞嗪、萘啶、喹喔啉、邻二氮萘、咔唑、菲啶、吖啶、吩嗪、噻唑、异噻唑、吩噻嗪、噁唑、异噁唑、噁二唑、吩噁嗪、2-,3-或4-吡啶基、2-,3-,4-,5-,或8-喹啉基、1-,3-,4-,或5-异喹啉基、1-,2-,或3-吲哚基、和2-,或3-噻吩基。所述基团可以是末端基团或桥接基团。
本发明使用的术语“杂原子”或其变体如“杂”是指O、N、NH和S。
本发明实施例公开的一些化合物可以以单一立体异构体、外消旋体、和/或对映异构体和/或非对映异构体混合物存在。所有此类单一立体异构体、外消旋体和其混合物均可认为在本发明描述或要求的主题范围之内。
此外,通式(I)在适用情况下,旨在包含所述化合物的溶剂化物以及非溶剂化形式。因此,通式(I)包括具有指明结构的化合物,包括其水合物或溶剂化物形式,以及其非水合物和非溶剂化物形式。
术语“药学上可接受的盐”是指在健全的医疗判断范围之内,适合用于接触人类或动物的组织而没有过度的毒性、刺激性、过敏反应等,并且与合理的效益/风险比率相应的那些盐。本领域技术人员周知,药学上可接受的盐由S.M.Berge等人在J.Pharmaceutical Sciences,1977,66:1-19中详细描述了药学上可接受的盐。所述的盐可以在最后分离纯化本发明化合物时就地制备得到,或分别通过游离碱与合适的有机酸反应制备得到。本发明化合物合适的药学上可接受的酸加成盐可以通过无机酸或有机酸制备得到。此类无机酸的实例有盐酸、氢溴酸、氢碘酸、硝酸、碳酸、硫酸、和磷酸。合适的有机酸可以选自脂肪族有机酸类、脂环族有机酸类、芳香族有机酸类、杂环有机酸类、羧基有机酸类、和磺酸有机酸类,有机酸的实例有甲酸、乙酸、丙酸、琥珀酸、乙醇酸、葡萄糖酸、乳酸、苹果酸、酒石酸、柠檬酸、抗坏血酸、葡萄糖醛酸、富马酸、顺丁烯二酸、丙酮酸、烷基磺酸、芳基磺酸、天冬氨酸、谷氨酸、苯甲酸、氨茴酸、甲磺酸、水杨酸、p-羟基苯甲酸、苯基乙酸、扁桃酸、阿波酮酸、帕莫酸、泛酸、对氨基苯磺酸、环己基氨基磺酸、硬脂酸、海藻酸、β-羟基酪酸、半乳糖二酸、和半乳糖醛酸。本发明化合物合适的药学上可接受的碱加成盐包括由锂、钠、钾、镁、钙、铝、和锌制成的金属盐,和由有机碱譬如胆碱、二乙醇胺、吗啉制成的有机盐。或者由N,N'-二苄基乙二胺、氯普鲁卡因、胆碱、二乙醇胺、乙二胺、葡甲胺(N-甲基葡糖胺)、普鲁卡因制成的有机盐,铵盐,季铵盐如四甲基铵盐,氨基酸加成盐如甘氨酸和精氨酸形成的盐。如果化合物均为固体,本领域技术人员应理解本发明化合物、药物和盐可以不同晶型或多晶型形式存在,其所有存在形式均可认为在本发明和指定通式的范围之内。
“前药”是指化合物在体内通过代谢方式(如通过水解、还原或氧化作用)可转换成本发明所述化合物。例如本发明包含羟基基团的化合物的酯类前药可以通过体内水解,从而转换成母分子。合适的酯类实例有醋酸酯类、柠檬酸酯类、乳酸酯类、酒石酸酯类、丙二酸酯类、草酸酯类、水杨酸酯类、丙酸酯类、琥珀酸酯类、富马酸酯类、马来酸酯类、亚甲基-双-β-羟基萘甲酸酯类、龙胆酸酯(gestisates)、羟乙基磺酸酯类、二-p-甲苯酰酒石酸酯类、甲烷磺酸酯类、乙烷磺酸酯类、苯磺酸酯类、p-甲苯磺酸酯类、环己基氨基磺酸酯类和奎尼酸酯类。
本发明所使用的术语“治疗”是指治愈性治疗和预防性治疗。因此,本发明上下文公开的术语“治疗”包含医治、改善减轻或调和癌症或其相关症状的严重程度。
“预防”或“防止”是指预防/防止所述癌症的发生或调和所述癌症的严重程度,如果所述癌症病情发展是继本发明化合物或药物组合物给药之后。此举可以预防临床上明显的有害细胞整体增殖的发作,或个人处于危险中有害细胞快速增殖的临床前明显阶段的发作。同样可认为恶性细胞转移的预防、或恶性细胞累进的捕捉或逆转均包含在所述这个定义中。
术语“治疗有效的”或“药理学上有效的”旨在证明每个药物的合格量,即通过每个药物本身的治疗,从而将实现改善疾病严重程度和发生频率的目标,同时避免常与其他疗法相关的严重副反应。
“药学上可接受的载体、稀释剂或赋形剂”包括但不限于,任何生理学上的缓冲(即pH值约为7.0-7.4)媒介包含合适的水溶性有机载体、常规溶剂、分散介质、填充剂、固体载体、包衣、抗菌剂和抗真菌剂、等渗剂和吸收延迟剂。合适的水溶性有机载体包括但不限于生理盐水、葡萄糖、玉米油、二甲亚砜和明胶胶囊。其他常规添加剂包括乳糖、甘露醇、玉米淀粉、马铃薯淀粉,粘合剂如结晶纤维素、纤维素衍生物、阿拉伯树胶、明胶,分解剂如羧甲基纤维素钠,和润滑剂如滑石粉或硬脂酸镁。
“患者”包括任何人类或非人类动物。因此,除了用于人类治疗之外,本发明化合物还可以用于兽医哺乳动物的治疗,包括宠物和家畜,譬如但不限于狗、猫、马、牛、羊和猪。
本发明上下文中的术语“给予”和其变体包括“施予”和“给药”,包括通过任何适当的方法,接触、应用、递送或提供本发明化合物或组合物至有机体或表面。
II.本发明化合物的合成
本发明涉及本发明通式(I)所示的功能化的吲哚类化合物,以及所述化合物作为治疗药物的用途。
通式(I)所示的化合物,或其盐、水合物或溶剂化物,可以通过本领域技术人员已知的方法来制备得到。对于制备式(I)化合物的一般合成方案均如下所述。
对于制备式(I)所示化合物,目前优选的合成路线的第一步是使吲哚结构骨架与若干连接基团的其中一个进行连接。化合物与C或O基团进行连接的具体应用条件如方案4所示。
下一步是N-烷基化所述取代的吲哚,如方案5所示。或者,所述N-烷基化可以在所述连接基团进行连接之前进行。
所述N-烷基化吲哚可以进一步与若干连接基团连接,N-连接化合物的具体应用条件如方案6所示。
上述方案4-6所述方法可提供一个或多个优点包括高产率、控制立体化学、合成步骤少、和对于放大生产而言,其反应条件温和。
上述方法仅仅是代表性的,对于本领域技术人员而言,路线的改变和变化均是显而易见的,并均在本发明公开的广泛的范围之内。
III.使用本发明化合物的治疗方法
本发明通式(I)所示化合物和其药物组合物,可以用于增殖性疾病的治疗或预防,尤其是癌症。本发明化合物和组合物可以用于多种多样癌症(肿瘤)的治疗,包括但不限于固体肿瘤,譬如乳腺癌、肺癌、前列腺癌、卵巢癌、子宫癌、脑癌、皮肤癌、结肠癌和膀胱癌。
有利地,本发明化合物可拥有优越的药学性质,如通过葡萄糖转移酶和其他水溶性转移酶如硫化酶(sulfases)对共轭结合提高抵抗,所述硫化酶(sulfases)可以对增殖细胞如癌症细胞过表达。这可能有助于赋予优越的药学性质,例如通过减少接合和消除而体现的增强的药代动力学特征。
适合本发明化合物递送的药物组合物及其制备方法,对于本领域技术人员而言是显而易见的。所述组合物及其制备方法可在如Remington’s Pharmaceutical Sciences,19th Edition(Mack Publishing Company,1995)中找到。
本发明化合物或药物组合物可以口服、静脉给药、滴鼻、直肠给药、不经肠道给药、皮下给药、肌内注射、外敷、或任何可递送活性药物有效量至组织或位点进行处理或治疗的方法。应领会不同的剂量可被要求用于不同病症的治疗。药物的有效量是指引起肿瘤细胞计数、生长或大小在统计学上显著减少的量。可对本发明药物响应的肿瘤疾病包括但不限于乳腺癌。
本发明化合物或药物组合物的剂型和用量可以很容易参照已知的治疗或预防方案来确定。
譬如,所述化合物和药物组合物可以配制成口服、注射、直肠、不经肠道、皮下、静脉或肌内递送的剂型。特殊剂型的非限制性实例包括片剂、胶囊、囊片、粉剂、颗粒剂、注射剂、安瓶剂、小瓶、随时可用的溶液剂或混悬剂、冻干材料、栓剂和埋植剂。所述固体剂型,如所述片剂或胶囊可含有任何数量的合适的药学上可接受的上述赋形剂或载体。
对于静脉给药、肌内给药、皮下给药或腹腔内给药,一个或多个化合物可以与无菌水溶液结合,其中无菌水溶液可与接受者的血液更好地等渗。所述制剂可以通过在水中溶解固体活性成分来制备得到,所述水中含有生理学上可相容的物质如氯化钠或甘氨酸,和一个与生理条件相容的缓冲pH体系,从而产生一种水溶液,并使所述溶液表现为无菌。所述制剂可以单位剂量或多剂量存在于包装容器中,如密封安瓶或小瓶。
对于使用本发明化合物和/或药物组合物治疗疾病状况,其给予治疗有效化合物的量和给药方案取决于多种因素,包括年龄,体重,性别,和患者医疗状况,疾病严重程度,给药途径和频率,特定化合物的使用,有害的增殖细胞的位置,以及个人治疗的药代动力学性质,因此可能相差很大。如果所述化合物是通过局部而非系统给药,并且用于预防而非用于治疗,药物剂量通常会更低。这种治疗方法可以根据需要经常给药,并且在一段时间内通过治疗医生来判断其必要性。本领域技术人员将领会,所述抑制剂的给药方案或治疗有效量在给药时需要针对每个个体进行优化。所述药物组合物可以含有活性成分的量为约0.1至2000mg的范围,优选约0.5至500mg的范围,最优选在约1和200mg之间。日剂量为约0.01至100mg/kg体重,优选在约0.1和约50mg/kg体重之间,可能是合适的。所述日剂量可以每天以1至4剂量进行给药。
本发明化合物可以连同上述药物载体、稀释剂或赋形剂进行给药。或者除此之外,所述化合物可以联合其他药物如化学治疗药物或免疫刺激药物或治疗药物进行给药。
术语“联合治疗”或“辅助治疗”定义为使用本发明化合物和一个或多个其他药物,意在包含每个药物顺序进行给药的方案,并将提供联合用药的有益效果,并且意在包含将这些药物实质上同时进行共同给药,如含有这些活性成分固定比率的单个制剂配方,或以多个、每个药物分开的制剂配方共同给药。
根据本发明各种实例,一个或多个通式(I)所示化合物可以联合一个或多个其他治疗药物进行配制或给药。因此,根据本发明各种实例,一个或多个通式(I)所示化合物可以包括在用外科手术和/或其他已知处理或治疗药物,例如其他抗癌药物,尤其是化学治疗药物、放射治疗药物、和/或辅助或预防用药物的联合治疗方案中。
已有大量抗肿瘤药物可通过商用、临床评价和临床前研究途径获得,并通过药物联合化学治疗,可选用作为癌症或其他肿瘤疾病的治疗。此类抗肿瘤药物分为几个大类,即抗生素类药物、烷化剂、抗代谢物、激素类药物、免疫类药物、干扰素类药物、和一类其他药物。或者,其他抗肿瘤药物,如间质金属结合蛋白酶抑制剂可被使用。可用于联合治疗的合适药物将通过本领域技术人员来识别。合适的药物已被列举,例如在默克索引An Encyclopaedia of Chemicals,Drugs and Biologicals,12th Ed.,1996中,其全部内容将并入本发明作为参考。
联合方案可以涉及活性药物在不同情况下一起、顺序、或以适当的定距离间隔进行给药。包含本发明化合物的活性药物的联合可以是协同作用。
所述通式(I)所示化合物的共同给药可以被作为化学治疗药物或其他抗肿瘤药物并处在同一单位剂量的通式(I)所示化合物所影响,或通式(I)所示化合物和化学治疗药物或其他抗肿瘤药物可以同时或在相似时间,存在于离散单位剂量给药的个体中。顺序给药可以是任何顺序要求,当第二个或后续化合物被给药时,尤其是当期望得到累积效应或协同效应时,可以要求第一个或初始化合物正在进行的生理效应是当前的。
本发明的实例将参照实施例来进行详细讨论,所述实施例将仅作为范例,其无论如何不应该被认为限制本发明的范围。
实施例
步骤1:2,3-二甲基-1H-吲哚-5-甲醛的制备
在-78℃下,向5-溴-2,3-二甲基-1H-吲哚(5.0g,22.42mmol)的干燥THF(50mL)搅拌溶液中,加入t-BuLi(44.8mL,67.20mmol)。所得反应混合物在相同温度下搅拌反应1小时。然后在-78℃下,向反应体系中加入干燥DMF(5.0mL,65.00mmol)。在所述温度下进一步搅拌反应2小时。待起始物料消耗完毕后,在-40℃下所述反应采用饱和氯化铵溶液淬灭,并且用乙酸乙酯萃取。合并的有机层用水和盐水洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物通过快速柱层析色谱,并使用20-25%乙酸乙酯的石油醚作为洗脱剂进行纯化,得到2,3-二甲基-1H-吲哚-5-甲醛为黄色固体(3.0g,77%)。LCMS:m/z 174.0[M+H]+。
步骤2和3:3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸甲酯的制备
室温下,将对甲苯磺酰肼(5.36g,28.9mmol)加至2,3-二甲基-1H-吲哚-5-甲醛(5.0g,28.9mmol)的干燥1,4-二氧六环(100mL)搅拌溶液中。在冷却至0℃之前,将温度升至80℃并搅拌反应2小时。
向所述粗品2,3-二甲基-5-((1-对甲苯磺酰基-2λ2-二氮烷基)甲基)-1H-吲哚的反应体系中,加入K2CO3(5.96g,43.2mmol)和(3-(甲氧基羰基)苯基)硼酸(5.18g,28.8mmol)。将反应温度升至110℃并搅拌反应4小时。待起始物料消耗完毕后,将反应体系浓缩,并用水稀释,然后用乙酸乙酯萃取。合并的有机层用水和盐水洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物通过快速柱层析色谱,并使用20-25%乙酸乙酯的石油醚作为洗脱剂进行纯化,得到3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸甲酯为棕色固体(4.0g,47%)。LCMS:m/z 294.38[M+H]+。
步骤4:3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸的制备
将3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸甲酯(4.0g,13.60mmol)溶于THF:H2O:MeOH(6:2:2)的混合物。0℃下,加入LiOH.H2O(1.14g,27.20mmol)。反应混合物允许在室温下搅拌反应16个小时。待起始物料消耗完毕后,将反应体系浓缩,然后加入乙酸乙酯和水,使其分层。收集水层,并在0℃下采用饱和柠檬酸溶液酸化。过滤,得到固体,并真空干燥,得到3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸为棕色固体(2.9g,77%)。LCMS:m/z 280.39[M+H]+。
步骤5:(3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮的制备
向3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸(300mg,1.08mmol)的DMF(5mL)搅拌溶液中加入DIPEA(0.5mL),然后搅拌10分钟,随后加入HATU(817.6mg,2.150mmol)并搅拌反应30分钟。将反应体系冷却至0℃,并加入1-(4-氟苯乙基)哌嗪(246.2mg,1.183mmol)。然后将反应混合物在室温下搅拌反应过夜。待起始物料消耗完毕后,将反应混合物倒入冰水中。过滤,收集所得沉淀,并干燥得到(3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(300mg,60%)。LCMS:m/z 470.23[M+H]+。通过此方法制备得到其他类似物:
3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(58%)。LCMS:m/z482.47[M+H]+。
(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)甲酮(56%)。LCMS:m/z 496.48[M+H]+。
(3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(70%)。LCMS:m/z 470.32[M+H]+。
3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮(72%)。LCMS:m/z 482.41[M+H]+。
3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-苯乙基哌嗪-1-基)甲酮(62%)。LCMS:m/z 452.23[M+H]+。
步骤6-1:(3-((2,3-二甲基-1-(3-氯丙基)-1H-吲哚-5-基)甲基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮的制备
0℃下,将NaH(30.6mg,1.2779mmol)分批加至(3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(300mg,0.6389mmol)的DMF(4mL)搅拌溶液中。反应混合物允许升至室温搅拌30分钟。然后在0℃下滴加溴氯丙烷(0.13mL,1.2779mmol),所得混合物在室温下搅拌反应3小时。待起始物料消耗完毕,向反应混合物中加入冰水,然后用乙酸乙酯萃取。所得有机层用盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物通过快速柱层析色谱,并使用乙酸乙酯作为洗脱剂进行纯化,得到(3-((2,3-二甲基-1-(3-氯丙基)-1H-吲哚-5-基)甲基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮为褐色粘性液体(200mg,57%)。LCMS:m/z 546.0[M+H]+。
通过此方法制备得到其他类似物:
(3-((2,3-二甲基-1-(3-氯丙基)-1H-吲哚-5-基)甲基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(50%)。LCMS:m/z 558.0[M+H]+。
(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)甲酮(58%)。LCMS:m/z 572.0[M+H]+。
(3-((2,3-二甲基-1-(3-氯丙基)-1H-吲哚-5-基)甲基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(43%)。LCMS:m/z546.39[M+H]+。
(3-((2,3-二甲基-1-(3-氯丙基)-1H-吲哚-5-基)甲基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮(43%)。LCMS:m/z 558.45[M+H]+。
(3-((2,3-二甲基-1-(3-氯丙基)-1H-吲哚-5-基)甲基)苯基)(4-苯乙基哌嗪-1-基)甲酮(45%)。LCMS:m/z 528.31[M+H]+。
步骤6-2:化合物3501,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮的制备
室温下,向(3-((2,3-二甲基-1-(3-氯丙基)-1H-吲哚-5-基)甲基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(200mg,0.366mmol)的乙腈(5mL)搅拌溶液中,加入碘化钠(137.1mg,0.9155mmol)和碳酸钠(116.4mg,1.0986mmol),随后加入N-甲基哌嗪(91.70mg,0.9155mmol)。将反应混合物加热至75℃搅拌反应16小时。待起始物料消耗完毕后,将反应混合物冷却至室温,并用乙酸乙酯(60mL)稀释,用水洗和盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物通过快速柱层析色谱,并使用5%甲醇-CH2Cl2作为洗脱剂进行纯化,得到(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(化合物3501)为淡黄色胶状固体(43mg,19%)。
1H NMR(300MHz,d6-DMSO):δ7.37–7.30(m,2H),7.28–7.22(m,4H),7.19–7.13(m,2H),7.09(t,J=9.0Hz,2H),6.91(br d,J=8.3Hz,1H),4.09–4.02(m,4H),3.66–3.46(m,4H),2.71(t,J=7.1Hz,2H),2.64–2.15(m,22H),2.14(s,3H),1.81–1.71(m,2H)。LCMS:m/z 610.56[M+H]+。
通过此方法制备得到其他类似物:
化合物3502,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(34%)。
1H NMR(400MHz,CD3OD):δ7.41–7.37(m,2H),7.25(br s,1H),7.23–7.19(m,2H),7.15–7.07(m,3H),6.92(br d,J=6.8Hz,1H),6.84(d,J=8.8Hz,2H),4.11(t,J=6.8Hz,2H),4.08(s,2H),3.76(s,3H),3.72(br s,2H),3.46(br s,2H),2.79–2.20(m,24H),2.18(s,3H),1.88(五重峰,J=6.8Hz,2H)。LCMS:m/z 622.58[M+H]+。
化合物3503,(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)甲酮(18%)。
1H NMR(400MHz,CD3OD):δ7.42–7.33(m,2H),7.27–7.22(m,2H),7.20(br d,J=6.8Hz,1H),7.14(br s,1H),6.92(dd,J=8.0Hz,1.2Hz,1H),6.83–6.77(m,2H),6.65(dd,J=8.4Hz,1.2Hz,1H),5.90(s,2H),4.13(t,J=6.8Hz,2H),4.08(s,2H),3.71(br s,2H),3.34(br s,2H),2.77(br s,4H),2.70–2.65(m,2H),2.59–2.47(m,11H),2.36(t,J=6.8Hz,2H),2.31(s,3H),2.25(br s,2H),2.18(s,3H),1.89(五重峰,J=6.9Hz,2H)。LCMS:m/z 636.54[M+H]+。
化合物3504,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(12%)。
1H NMR(300MHz,CD3OD):δ7.40–7.34(m,2H),7.33–7.18(m,4H),7.15(br s,1H),7.06–6.96(m,2H),6.96–6.87(m,2H),4.12–4.08(m,4H),3.71(br s,2H),3.38(br s,2H),2.81–2.71(m,2H),2.71–2.47(m,10H),2.46–2.21(m,12H),2.32(s,3H),1.87(五重峰,J=6.9Hz,2H)。LCMS:m/z 610.6[M+H]+。
化合物3505,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮(12%)。
1H NMR(300MHz,d6-DMSO):δ7.35–7.32(m,2H),7.28–7.25(m,2H),7.21–7.13(m,3H),6.91(br d,J=8.4Hz,1H),6.82–6.72(m,3H),4.09–4.03(m,4H),3.73(s,3H),3.53(br s,2H),3.25(br s,2H),2.74–2.61(m,4H),2.47–2.15(m,20H),2.14(s,3H),1.82–1.71(m,2H)。LCMS:m/z 622.58[M+H]+。
化合物3506,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-苯乙基哌嗪-1-基)甲酮(12%)。
1H NMR(300MHz,d6-DMSO):δ7.42–7.08(m,11H),6.94–6.89(m,1H),4.11–4.01(m,4H),3.62–3.43(m,4H),2.76–2.67(m,2H),2.63–2.10(m,25H),1.91–1.81(m,2H)。LCMS:m/z 592.58[M+H]+。
方案8.化合物3507–3512的一般合成
步骤1:5-甲氧基-2,3-二甲基-1H-吲哚的制备
将2-丁酮(11.93mL,128.8mmol)加至4-甲氧基肼盐酸盐(15.00g,85.89mmol)的乙酸(150mL)搅拌溶液中,然后将反应混合物加热至80℃搅拌反应1.5小时。待起始物料消耗完毕后,通过旋转蒸发仪除去乙酸,反应体系采用饱和NaHCO3溶液碱化。过滤,收集所得灰色沉淀,并干燥1小时。所得粗产物通过快速柱层析色谱,并使用20%乙酸乙酯的石油醚作为洗脱剂进行纯化,得到5-甲氧基-2,3-二甲基-1H-吲哚为灰色固体(8.8g,59%)。LCMS:m/z 176.23[M+H]+。
步骤2:2,3-二甲基-1H-吲哚-5-醇的制备
0℃下,将BBr3(12.18mL,128.40mmol)加至5-甲氧基-2,3-二甲基-1H-吲哚的DCM(50mL)搅拌溶液中。将温度保持在0-5℃搅拌反应2小时。待起始物料消耗完毕后,反应混合物采用饱和NaHCO3碱化,然后用CH2Cl2萃取。所得有机层用盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物通过快速柱层析色谱,并使用20-50%乙酸乙酯的石油醚作为洗脱剂进行纯化,得到2,3-二甲基-1H-吲哚-5-醇为米白色固体(8.2g,100%)。LCMS:m/z 162.08[M+H]+。
步骤3:5-羟基-2,3-二甲基-1H-吲哚-1-羧酸叔丁酯的制备
室温下,向2,3-二甲基-1H-吲哚-5-醇(7.20g,44.7mmol)的乙腈(72mL)搅拌溶液中加入Boc-酸酐(29.2g,134mmol)和DMAP(0.55g,4.472mmol)。反应体系在室温下搅拌过夜。待起始物料消耗完毕后,减压蒸去乙腈,得到N-Boc-5-羟基吲哚和N,O-二-Boc-保护的化合物(8.2g,51.42mmol)的粗品混合物。将所述混合物再次溶于甲醇(828mL),加入K2CO3(21.3g,154.2mmol),所得混合物在室温下搅拌反应2小时。反应完毕后,将反应混合物冷却至0℃,然后加入乙酸(10mL),所得混合物搅拌10分钟。反应体系采用乙酸乙酯萃取。所得有机层用水洗和盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经100-200目硅胶柱层析,并采用20%乙酸乙酯的石油醚进行洗脱纯化,得到5-羟基-2,3-二甲基-1H-吲哚-1-羧酸叔丁酯为棕色液体(9.5g,72%)。LCMS:m/z 262.40[M+H]+。
步骤4:5-(3-(甲氧基羰基)苯氧基)-2,3-二甲基-1H-吲哚-1-羧酸叔丁酯的制备
向5-羟基-2,3-二甲基-1H-吲哚-1-羧酸叔丁酯(7.00g,26.7mmol)的DCM(100mL)搅拌溶液中加入(3-(甲氧基羰基)苯基)硼酸(14.4g,80.361mmol)。然后加入Cu(OAc)2(12.16g,66.96mmol),随后再加入NEt3(18.5ml,133.93mmol),反应体系通氧气净化4小时。整个反应体系在氧气氛下搅拌反应过夜。待起始物料消耗完毕后,反应混合物通过硅藻土过虑。滤液用水稀释,并采用DCM萃取。所得有机层用盐水洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经100-200目硅胶柱层析,并采用10%乙酸乙酯的石油醚进行洗脱纯化,得到5-(3-(甲氧基羰基)苯氧基)-2,3-二甲基-1H-吲哚-1-羧酸叔丁酯为棕色液体(8.2g,77%)。LCMS:m/z 396.43[M+H]+。
步骤5:3-((2,3-二甲基-1H-吲哚-5-基)氧基)苯甲酸的制备
向5-(3-(甲氧基羰基)苯氧基)-3-甲基-1H-吲哚-1-羧酸叔丁酯(8.20g,20.8mmol)的THF(100mL)和水(100mL)搅拌溶液中,加入LiOH.H2O(17.4g,415mmol)。反应混合物在室温下搅拌反应4小时。待起始物料消耗完毕后,减压蒸去THF,并将反应体系冷却至0℃,采用1N HCl酸化(至pH 1),然后用乙酸乙酯萃取。所得有机层用无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物与正戊烷一起研磨,得到纯的3-((2,3-二甲基-1H-吲哚-5-基)氧基)苯甲酸为米白色固体(5.0g,86%)。LCMS:m/z 282.2[M+H]+。
步骤6:(3-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮的制备
向3-((2,3-二甲基-1H-吲哚-5-基)氧基)苯甲酸(0.25g,0.88mmol)的DMF(3mL)搅拌溶液中加入DIPEA(0.70mL,4.44mmol)。搅拌10分钟后,加入HATU(0.50g,1.33mmol),所得混合物在室温下继续搅拌反应30分钟。将反应体系冷却至0℃,并加入1-(4-氟苯乙基)哌嗪(0.32g,1.33mmol),反应混合物在室温下搅拌反应过夜。待起始物料消耗完毕后,将反应混合物倒入冰水中,并用乙酸乙酯萃取。所得有机层用水洗和盐水洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经100-200目硅胶柱层析,并采用30%乙酸乙酯的石油醚进行洗脱纯化,得到(3-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮为黄色固体(410mg,97%)。LCMS:m/z 472.52[M+H]+。
通过此方法制备得到其他类似物:
(3-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(92%)。LCMS:m/z 484.56[M+H]+。
(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(3-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)甲酮(97%)。LCMS:m/z 498.50[M+H]+。
(3-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(71%)。LCMS:m/z 472.55[M+H]+。
(3-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮(92%)。LCMS:m/z 484.56[M+H]+。
(3-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-苯乙基哌嗪-1-基)甲酮(89%)。LCMS:m/z 454.47[M+H]+。
步骤7-1:(3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮的制备
0℃下,将KOtBu(0.29g,4.434mmol)分批加至(3-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(0.41g,0.88mmol)的DMF(5mL)搅拌溶液中。反应混合物允许升至室温搅拌反应30分钟。然后在0℃下,向反应混合物中滴加溴氯丙烷(0.43mL,4.43mmol)。将反应混合物升至室温搅拌反应3小时。待起始物料消耗完毕后,向反应混合物中加入冰水,并用乙酸乙酯萃取。所得有机层用盐水溶液洗,无水Na2SO4干燥,并减压浓缩得到粗产物。所得粗产物经快速柱层析色谱,并使用5%乙酸乙酯的石油醚作为洗脱剂进行纯化,得到(3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮为棕色胶状固体(130mg,27%)。LCMS:m/z 548.59[M+H]+。
通过此方法制备得到其他类似物:
(3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(64%)。LCMS:m/z 560.53[M+H]+。
(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)甲酮(74%)。LCMS:m/z 574.89[M+H]+。
(3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(39%)。LCMS:m/z548.55[M+H]+。
(3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮(57%)。LCMS:m/z 560.53[M+H]+。
(3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-苯乙基哌嗪-1-基)甲酮(43%)。LCMS:m/z 530.41[M+H]+。
步骤7-2:化合物3507,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮的制备
室温下,向(3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(250mg,0.455mmol)的乙腈(10mL)搅拌溶液中,加入碘化钠(170mg,1.137mmol)和碳酸钠(241mg,2.27mmol),随后加入N-甲基哌嗪(182mg,1.82mmol)。将反应混合物加热至75℃并搅拌反应16小时。待起始物料消耗完毕后,将反应混合物冷却至室温,用乙酸乙酯(30mL)稀释,并用水洗和盐水洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析色谱,并使用5%甲醇-CH2Cl2作为洗脱剂进行纯化,得到(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(化合物3507)为米白色固体(87mg,31%)。
1H NMR(300MHz,d6-DMSO):δ7.42(d,J=9.0Hz,1H),7.38(t,J=7.8Hz,1H),7.24(dd,J=8.4Hz,6.0Hz,2H),7.12–7.06(m,3H),7.02–6.96(m,2H),6.81(dd,J=8.7Hz,2.1Hz,1H),6.76(br s,1H),4.12(t,J=6.9Hz,2H),3.57(br s,2H),3.33(br s,2H),2.72–2.14(m,26H),2.13(s,3H),1.85–1.74(m,2H)。LCMS:m/z 621.54[M+H]+。
通过此方法制备得到其他类似物:
化合物3508,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(6%)。
1H NMR(300MHz,d6-DMSO):δ7.43(d,J=8.7Hz,1H),7.38(t,J=7.5Hz,1H),7.14–7.08(m,3H),7.04–6.96(m,2H),6.86–6.79(m,3H),6.77(br s,1H),4.12(t,J=6.9Hz,2H),3.71(s,3H),3.51(br s,4H),2.70–2.15(m,24H),2.13(s,3H),1.88–1.74(m,2H)。LCMS:m/z 624.65[M+H]+。
化合物3509,(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)甲酮(35%)。
1H NMR(300MHz,CD3OD):δ7.41–7.33(m,2H),7.15–7.00(m,3H),6.85–6.79(m,2H),6.73–6.60(m,2H),6.65(br d,J=8.1Hz,1H),5.89(s,2H),4.16(t,J=6.9Hz,2H),3.81–3.51(m,2H),3.53–3.37(m,2H),2.72–2.64(m,2H),2.61–2.42(m,10H),2.42–2.25(m,12H),2.17(s,3H),1.94(五重峰,J=6.9Hz,2H)。LCMS:m/z 638.48[M+H]+。
化合物3510,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(41%)。
1H NMR(300MHz,CD3OD):δ7.42–7.32(m,2H),7.28(td,J=7.8Hz,6.0Hz,1H),7.09(d,J=2.4Hz,1H),7.07–6.87(m,5H),6.84–6.78(m,2H),4.17(t,J=6.9Hz,2H),3.70(br s,2H),3.43(br s,2H),2.83–2.77(m,2H),2.64–2.32(m,19H),2.31(s,3H),2.17(s,3H),1.91(五重峰,J=6.9Hz,2H)。LCMS:m/z 612.51[M+H]+。
化合物3511,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮(15%)。
1H NMR(300MHz,d6-DMSO):δ77.38(t,J=7.5Hz,1H),7.39(d,J=8.7Hz,1H),7.18(dd,J=8.4Hz,6.6Hz,1H),7.09(d,J=2.1Hz,1H),7.08–7.00(m,2H),6.83–6.72(m,5H),4.16(t,J=6.9Hz,2H),3.77(s,3H),3.71(br s,2H),3.43(br s,2H),2.73(dd,J=9.6Hz,6.6Hz,2H),2.63–2.29(m,19H),2.28(s,3H),2.17(s,3H),1.90(五重峰,J=7.2Hz,2H)。LCMS:m/z 624.49[M+H]+。
化合物3512,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-苯乙基哌嗪-1-基)甲酮(60%)。
1H NMR(300MHz,d6-DMSO):δ7.43(d,J=8.7HZ,1H),7.38(t,J=7.5Hz,1H),7.32–7.16(m,5H),7.13(d,J=2.1Hz,1H),7.03–6.96(m,2H),6.82(dd,J=8.4Hz,2.4Hz,1H),6.77(br s,1H),4.12(t,J=6.9Hz,2H),3.54(br s,4H),3.03–2.18(m,24H),2.14(s,3H),1.88–1.64(m,2H)。LCMS:m/z 594.52[M+H]+。
方案9.化合物3513–3518的一般合成
步骤1:(3-硝基苯基)(4-苯乙基哌嗪-1-基)甲酮的制备
向3-硝基苯甲酸(1.0g,5.9mmol)的DMF(10mL)搅拌溶液中加入DIPEA(1.97mL,11.3mmol)。搅拌10分钟后,加入HATU(4.55g,11.97mmol),反应混合物在室温下进一步搅拌30分钟。将反应体系冷却至0℃,然后加入1-苯乙基哌嗪(1.1mL,5.8mmol),反应混合物在室温下搅拌反应3小时。反应进度通过TCL监测。待起始物料消耗完毕后,将反应混合物倒入冰水中,并用乙酸乙酯萃取。所得有机层用水洗和盐水洗,无水Na2SO4干燥,减压浓缩。所得粗产物经快速柱层析色谱,并使用5%甲醇-CH2Cl2作为洗脱剂进行纯化,得到(3-硝基苯基)(4-苯乙基哌嗪-1-基)甲酮为棕色固体(1.5g,74%)。
通过此方法制备得到其他类似物:
(4-(4-氟苯乙基)哌嗪-1-基)(3-硝基苯基)甲酮(94%)
(4-(4-甲氧基苯乙基)哌嗪-1-基)(3-硝基苯基)甲酮(70%)
(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(3-硝基苯基)甲酮(31%)
(4-(3-氟苯乙基)哌嗪-1-基)(3-硝基苯基)甲酮(64%)
(4-(3-甲氧基苯乙基)哌嗪-1-基)(3-硝基苯基)甲酮(49%)
步骤2:(3-氨基苯基)(4-苯乙基哌嗪-1-基)甲酮的制备
室温下,向(3-硝基苯基)(4-苯乙基哌嗪-1-基)甲酮(1.50g,4.42mmol)的乙醇和水(1:1,各15mL)搅拌溶液中,加入Fe粉(1.23g,22.1mmol)和NH4Cl(475mg,8.88mmol)。将反应混合物加热至60℃搅拌反应3小时。待起始物料消耗完毕后,将反应混合物通过硅藻土过滤,并蒸去乙醇。水层用乙酸乙酯萃取,所得有机层用盐水洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析色谱,并使用6%甲醇-CH2Cl2作为洗脱剂进行纯化,得到(3-氨基苯基)(4-苯乙基哌嗪-1-基)甲酮为棕色固体(1.0g,73%)。
通过此方法制备得到其他类似物:
(3-氨基苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(70%)
(3-氨基苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(45%)
(3-氨基苯基)(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)甲酮(41%)
(3-氨基苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(89%)
(3-氨基苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮(52%)
步骤3:5-溴-1-(3-氯丙基)-2,3-二甲基-1H-吲哚的制备
0℃下,将NaH(1.80g,44.6mmol)分批加至5-溴-2,3-二甲基-1H-吲哚(5.00g,22.3mmol)的DMF(50mL)搅拌溶液中。将混合物升至室温,并搅拌30分钟。0℃下,向反应体系中滴加溴氯丙烷(11.68mL,111.6mmol),反应混合物在室温下搅拌反应3小时。待起始物料消耗完毕后,向反应混合物中加入冰水,并用乙酸乙酯萃取。所得有机层用盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析色谱,并使用5%乙酸乙酯的石油醚作为洗脱剂进行纯化,得到5-溴-1-(3-氯丙基)-2,3-二甲基-1H-吲哚为粉色固体(2.6g,40%)。LCMS:m/z 302.10[M+H]+。
步骤4:5-溴-2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚的制备
室温下,向5-溴-1-(3-氯丙基)-2,3-二甲基-1H-吲哚(9.00g,29.9mmol)的乙腈(20mL)搅拌溶液中,加入碘化钠(11.2g,74.7mmol)、碳酸钠(7.93g,74.7mmol),然后加入N-甲基哌嗪(7.40g,74.7mmol)。将反应混合物加热至75℃搅拌反应16小时。待起始物料消耗完毕后,将反应混合物冷却至室温,并用乙酸乙酯(60mL)稀释,然后用水洗和盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析色谱,并使用5%甲醇-CH2Cl2作为洗脱剂进行纯化,得到5-溴-2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚为米白色固体(3.2g,30%)。LCMS:m/z 365.98[M+H]+。
步骤5:化合物3513,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮的制备
将(3-氨基苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(120mg,0.355mmol)和NaOtBu(78mg,0.82mmol)加至5-溴-2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚(100mg,0.273mmol)的1,4-二氧六环(3mL)搅拌溶液中。反应混合物通入氩气10分钟。然后向反应体系中加入Pd2(dba)3(17mg,0.019mmol)和2-二环己膦基-2’-(N,N-二甲胺)-联苯(16mg,0.041mmol),并再次通入氩气10分钟。将反应混合物加热至90℃搅拌反应16小时。待起始物料消耗完毕后,反应混合物用乙酸乙酯稀释,并用硅藻土过滤。所得有机层用水洗和盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物采用制备TCL,并使用5%甲醇的CH2Cl2洗脱纯化,得到(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(化合物3513)为淡黄色固体(50mg,15%)。
1H NMR(300MHz,CD3OD):δ7.29(d,J=8.4Hz,1H),7.26–7.12(m,4H),7.05–6.95(m,3H),6.93(dd,J=8.7Hz,1.8Hz,1H),6.82(br s,1H),6.67(br d,J=7.2Hz,1H),4.16(t,J=6.6Hz,2H),3.72(br s,2H),3.51(br s,2H),2.89–2.66(m,6H),2.66–2.42(m,13H),2.42–2.24(m,5H),2.18(s,3H),1.92(五重峰,J=6.9Hz,2H)。LCMS:m/z 611[M+H]+。
通过此方法制备得到其他类似物:
化合物3514,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(43%)。
1H NMR(400MHz,d6-DMSO):δ7.88(br s,1H),7.32(d,J=8.8Hz,1H),7.16(t,J=8.0Hz,1H),7.13–7.09(m,3H),6.91(br d,J=8.0Hz,1H),6.86(dd,J=8.8Hz,2.0Hz,1H),6.83(d,J=8.4Hz,2H),6.60(d,J=7.6Hz,1H),4.08(t,J=6.8Hz,2H),3.71(s,3H),3.52(br s,4H),2.66(dd,J=8.8Hz,6.8Hz,2H),2.46–2.11(m,22H),1.78(五重峰,J=6.8Hz,2H)。LCMS:m/z 623.17[M+H]+。
化合物3515,(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)甲酮(28%)。
1H NMR(300MHz,d6-DMSO):δ7.91(br s,1H),7.32(d,J=8.7Hz,1H),7.17(t,J=8.1Hz,1H),7.13(d,J=1.5Hz,1H),6.91(br d,J=8.4Hz,1H),6.86(dd,J=8.4Hz,1.5Hz,1H),6.83–6.75(m,3H),6.66(dd,J=8.1Hz,1.5Hz,1H),6.60(br d,J=7.2Hz,1H),5.95(s,2H),4.08(t,J=6.9Hz,2H),3.51(br s,4H),2.69–2.59(m,2H),2.48–2.14(m,22H),2.13(s,3H),1.82–1.72(m,2H)。LCMS:m/z 637.49[M+H]+。
化合物3516,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(22%)。
1H NMR(400MHz,d6-DMSO):δ7.90(br s,1H),7.35–7.27(m,2H),7.17(t,J=8.0Hz,1H),7.13(d,J=1.6Hz,1H),7.11–7.04(m,2H),7.00(td,J=8.4Hz,2.0Hz,1H),6.91(br d,J=8.0Hz,1H),6.87(dd,J=8.4Hz,2.1Hz,1H),6.80(br s,1H),6.60(br d,J=7.6Hz,1H),4.08(t,J=7.2Hz,2H),3.51(br s,4H),2.75(dd,J=8.4Hz,7.2Hz,2H),2.58–2.52(m,2H),2.49–2.16(m,20H),2.13(s,3H),1.81–1.72(m,2H)。LCMS:m/z 611.18[M+H]+。
化合物3517,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮(21%)。
1H NMR(400MHz,d6-DMSO):δ7.91(br s,1H),7.32(d,J=8.4Hz,1H),7.20–7.14(m,2H),7.13(d,J=2.0Hz,1H),6.91(dd,J=8.4Hz,1.2Hz,1H),6.86(dd,J=8.8Hz,2.0Hz,1H),6.71–6.62(m,4H),6.60(br d,J=7.2Hz,1H),4.08(t,J=6.8Hz,2H),3.73(s,3H),3.54(br s,4H),2.73–2.68(m,2H),2.53–2.15(m,22H),2.13(s,3H),1.82–1.72(m,2H)。LCMS:m/z 623.1[M+H]+。
化合物3518,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-苯乙基哌嗪-1-基)甲酮(13%)。
1H NMR(400MHz,d6-DMSO):δ7.90(br s,1H),7.32(d,J=8.4Hz,1H),7.30–7.11(m,7H),6.92(br d,J=7.2Hz,1H),6.87(br s,J=8.4Hz,1H),6.81(br s,1H),6.60(br d,J=7.2Hz,1H),4.09(t,J=6.8Hz,2H),3.52(br s,4H),2.75–2.68(m,2H),2.63–2.14(m,22H),2.13(s,3H),1.84–1.73(m,2H)。LCMS:m/z 593.55[M+H]+。
方案10.化合物3519–3524的一般合成
步骤1和2:4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸甲酯的制备
室温下,将对甲苯磺酰肼(2.14g,11.50mmol)加至2,3-二甲基-1H-吲哚-5-甲醛(2.0g,11.50mmol)的干燥1,4-二氧六环(50mL)搅拌溶液中。将温度升至80℃并保持2小时。
80℃下,向粗品2,3-二甲基-5-((1-对甲苯磺酰基-2λ2-二氮烷基)甲基)-1H-吲哚的反应体系中,加入K2CO3(2.38g,17.20mmol)和(4-(甲氧基羰基)苯基)硼酸(2.07g,11.50mmol)。然后将反应温度升至110℃并搅拌反应4小时。待起始物料消耗完毕后,将反应混合物浓缩,用水稀释,然后用乙酸乙酯萃取。合并的有机层用水洗和盐水洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析色谱,并使用20-25%乙酸乙酯的石油醚作为洗脱剂进行纯化,得到4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸甲酯为棕色固体(1.5g,45%)。LCMS:m/z 294.41[M+H]+。
步骤3:4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸的制备
0℃下,向4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸甲酯(3.0g,10.20mmol)的THF:H2O:MeOH(6:2:2)混合溶液中加入LiOH.H2O(1.28g,30.70mmol)。反应混合物在室温下搅拌反应16小时。待起始物料消耗完毕后,将反应混合物浓缩,然后加入乙酸乙酯和水,使其分层。收集水层,并在0℃下用饱和柠檬酸溶液酸化。过滤,收集所析出沉淀,真空干燥,得到4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸为棕色固体(1.8g,63%)。LCMS:m/z 280.39[M+H]+。
步骤4:(4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-苯乙基哌嗪-1-基)甲酮的制备
向4-((2,3-二甲基-1H-吲哚-5-基)氧基)苯甲酸(250mg,0.896mmol)的DMF(5mL)搅拌溶液中,加入DIPEA(0.5mL)。搅拌10分钟后,加入HATU(511.0mg,1.3440mmol),反应混合物继续搅拌30分钟。将反应体系冷却至0℃,加入1-苯乙基哌嗪(187.5mg,0.9856mmol),反应混合物在室温下搅拌反应过夜。待起始物料消耗完毕后,将反应混合物倒入冰水中。析出沉淀,过滤收集所得沉淀,并干燥,得到(4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-苯乙基哌嗪-1-基)甲酮为米白色固体(300mg,74%)。LCMS:m/z 452.34[M+H]+。
通过此方法制备得到其他类似物:
(4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(74%)。LCMS:m/z 470.1[M+H]+。
(4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(70%)。LCMS:m/z 482.0[M+H]+。
(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)甲酮(79%)。LCMS:m/z 496.0[M+H]+。
(4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(65%)。LCMS:m/z 470.32[M+H]+。
(4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮(67%)。LCMS:m/z 482.28[M+H]+。
步骤5-1:(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-苯乙基哌嗪-1-基)甲酮的制备
0℃下,将NaH(21.2mg,0.8857mmol)分批加至(4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-苯乙基哌嗪-1-基)甲酮(200mg,0.4428mmol)的DMF(4mL)搅拌溶液中。将混合物升至室温搅拌30分钟。0℃下,向反应体系中滴加溴氯丙烷(0.10mL,0.8857mmol),反应混合物在室温下搅拌反应3小时。待起始物料消耗完毕后,将冰水加至反应混合物中,然后用乙酸乙酯萃取。有机层用盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析色谱,并使用乙酸乙酯作为洗脱剂进行纯化,得到(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-苯乙基哌嗪-1-基)甲酮为棕色胶状固体(150mg,64%)。LCMS:m/z 528.34[M+H]+。
通过此方法制备得到其他类似物:
4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(57%)。LCMS:m/z 546.33[M+H]+。
4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(65%)。LCMS:m/z 558.41[M+H]+。
(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)甲酮(43%)。LCMS:m/z 572.34[M+H]+。
4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(65%)。LCMS:m/z 546.33[M+H]+。
4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(78%)。LCMS:m/z 558.36[M+H]+。
步骤5-2:化合物3524,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-苯乙基哌嗪-1-基)甲酮的制备
室温下,向(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-苯乙基哌嗪-1-基)甲酮(150mg,0.284mmol)的乙腈(5mL)搅拌溶液中,加入碘化钠(85.1mg,0.568mmol)和碳酸钠(90.3mg,0.852mmol),随后加入N-甲基哌嗪(71.1mg,0.710mmol)。将反应混合物加热至75℃搅拌反应16小时。待起始物料消耗完毕后,将反应混合物冷却至室温,用乙酸乙酯(40mL)稀释,并用水洗和盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析色谱,并使用5%甲醇-CH2Cl2作为洗脱剂进行纯化,得到(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-苯乙基哌嗪-1-基)甲酮(化合物3524)为米白色固体(37mg,22%)。
1H NMR(300MHz,d6-DMSO):δ7.36–7.12(m,11H),6.91(br d,J=8.7Hz,1H),4.07(t,J=7.2Hz,2H),4.01(s,2H),3.52(br s,4H),2.75–2.69(m,2H),2.61–2.56(m,2H),2.47–2.15(m,20H),2.14(s,3H),1.82–1.69(m,2H)。LCMS:m/z 592.58[M+H]+。
通过此方法制备得到其他类似物:
化合物3519,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(6%)。
1H NMR(300MHz,CD3OD):δ7.36(br s,4H),7.29–7.22(m,4H),7.03(t,J=9.0Hz,2H),6.94(br d,J=8.4Hz,1H),4.19(t,J=6.6Hz,2H),4.08(s,2H),3.71(br s,4H),3.20–2.64(m,18H),2.36(br s,6H),2.19(s,3H),1.99–1.88(m,2H)。LCMS:m/z 610.56[M+H]+。
化合物3520,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(23%)。
1H NMR(300MHz,CD3OD):δ7.32–7.23(m,6H),7.12(d,J=8.4Hz,2H),6.92(br d,J=8.7Hz,1H),6.82(d,J=8.4Hz,2H),4.07(t,J=6.6Hz,2H),4.01(br s,2H),3.70(s,3H),3.52(br s,4H),3.28–3.21(m,2H),2.52–2.18(m,22H),2.14(s,3H),1.82–1.71(m,2H)。LCMS:m/z 622.54[M+H]+。
化合物3521,(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)甲酮(18%)。
1H NMR(300MHz,CD3OD):δ7.32(br s,4H),7.27–7.20(m,2H),6.93(dd,J=9.0,2.1Hz,1H),6.73–6.63(m,3H),5.88(s,2H),4.17(t,J=6.9Hz,2H),4.07(s,2H),3.73(br s,4H),2.99–2.34(m,24H),2.19(s,3H),1.96-1.87(m,2H)。LCMS:m/z 636.54[M+H]+。
化合物3522,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(15%)。
1H NMR(400MHz,CD3OD):δ7.35–7.23(m,7H),7.11–7.04(m,2H),7.04-6.95(m,1H),6.91(br d,J=8.4Hz,2H),4.07(t,J=6.9Hz,2H),4.01(s,2H),3.51(br s,4H),2.79–2.68(m,2H),2.57–2.16(m,22H),2.14(s,3H),1.82–1.71(m,2H)。LCMS:m/z 610.53[M+H]+。
化合物3523,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮(化合物23)(20%)。
1H NMR(400MHz,CD3OD):δ7.33–7.22(m,6H),7.17(t,J=8.1Hz,1H),6.92(br d,J=7.8Hz,1H),6.81–6.72(m,3H),4.16–4.01(m,4H),3.76(s,3H),3.53(br s,4H),2.82–2.20(m,21H),2.18(s,3H),1.97(s,3H),1.81–1.68(m,2H)。LCMS:m/z 622.54[M+H]+。
方案11.化合物3525–3530的一般合成
步骤1:5-(4-(甲氧基羰基)苯氧基)-2,3-二甲基-1H-吲哚-1-羧酸叔丁酯的制备
向5-羟基-2,3-二甲基-1H-吲哚-1-羧酸叔丁酯(6.80g,26.0mmol)的CH2Cl2(70mL)搅拌溶液中,加入(4-(甲氧基羰基)苯基)硼酸(14.0g,78.1mmol),随后加入Cu(OAc)2(11.8g,65.1mmol)和TEA(34.0mL,260mmol)。向所述体系中通入氧气4小时进行净化。整个反应体系在氧气氛下搅拌反应过夜。待起始物料消耗完毕后,将反应混合物通过硅藻土滤筛过滤。滤液用水稀释,并用CH2Cl2萃取。所得有机层用盐水洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经100-200目硅胶柱层析,并使用10%乙酸乙酯的石油醚进行洗脱纯化,得到目标产品5-(4-(甲氧基羰基)苯氧基)-2,3-二甲基-1H-吲哚-1-羧酸叔丁酯为棕色液体(6.50g,63%)。LCMS:m/z 396.3[M+H]+。
步骤2:4-((2,3-二甲基-1H-吲哚-5-基)氧基)苯甲酸的制备
向5-(4-(甲氧基羰基)苯氧基)-2,3-二甲基-1H-吲哚-1-羧酸叔丁酯(6.50g,16.5mmol)的THF(75mL)、水(75mL)、和甲醇(75mL)搅拌溶液中,加入LiOH.H2O(13.8g,329mmol)。反应混合物在室温下搅拌反应4小时。待起始物料消耗完毕后,减压蒸去THF。将反应体系冷却至0℃,用1N HCl酸化(pH 1),然后用乙酸乙酯萃取。所得有机层用无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物与正戊烷一起研磨,得到纯的4-((2,3-二甲基-1H-吲哚-5-基)氧基)苯甲酸(4.00g,86%)。LCMS:m/z 282.34[M+H]+。
步骤3:(4-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮的制备
向4-((2,3-二甲基-1H-吲哚-5-基)氧基)苯甲酸(300mg,1.06mmol)的DMF(3mL)搅拌溶液中,加入DIPEA(0.93mL,5.33mmol)。搅拌10分钟后,加入HATU(0.6g,1.59mmol),然后反应混合物在室温下继续搅拌30分钟。将反应体系冷却至0℃,加入1-(4-氟苯乙基)哌嗪(0.40g,1.59mmol),反应混合物在室温下搅拌反应过夜。待起始物料消耗完毕后,将反应混合物倒入冰水中,用乙酸乙酯萃取。所得有机层用水洗和盐水洗,无水Na2SO4干燥,并减压浓缩得到粗产物。所得粗产物经100-200目硅胶柱层析,并使用30%乙酸乙酯的石油醚进行洗脱纯化,得到(4-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮为粘性棕色固体(450mg,89%)。LCMS:m/z 472.52[M+H]+。
通过此方法制备得到其他类似物:
(4-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(77%)。LCMS:m/z 484.50[M+H]+。
((4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(4-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)甲酮(87%)。LCMS:m/z 498.56[M+H]+。
(4-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(22%)。LCMS:m/z 472.52[M+H]+。
(4-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮(99%)。LCMS:m/z 484.56[M+H]+。
(4-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-苯乙基哌嗪-1-基)甲酮(23%)。LCMS:m/z 454.53[M+H]+。
步骤4-1:(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮的制备
0℃下,将NaH(75.0mg,1.88mmol)分批加至(4-((2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(445mg,0.944mmol)的DMF(5mL)搅拌溶液中。将混合物升至室温搅拌30分钟。0℃下,向反应混合物中滴加溴氯丙烷(0.19mL,1.88mmol)。反应混合物在室温下搅拌反应3小时。待起始物料消耗完毕后,加入冰水,反应混合物用乙酸乙酯萃取。所得有机层用盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析色谱,并使用5%乙酸乙酯的石油醚作为洗脱剂进行纯化,得到(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮为棕色胶状固体(340mg,66%)。LCMS:m/z 548.52[M+H]+。
通过此方法制备得到其他类似物:
(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(62%)。LCMS:m/z 560.93[M+H]+。
(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)甲酮(61%)。LCMS:m/z 574.92[M+H]+。
(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(62%)。LCMS:m/z548.32[M+H]+。
(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮(60%)。LCMS:m/z 560.93[M+H]+。
(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-苯乙基哌嗪-1-基)甲酮(67%)。LCMS:m/z 530.57[M+H]+。
步骤4-2:化合物3525,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮的制备
室温下,向(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(335mg,0.610mmol)的乙腈(4mL)搅拌溶液中,加入碘化钠(229mg,1.52mmol)和碳酸钠(194mg,1.83mmol),随后加入N-甲基哌嗪(153mg,1.52mmol)。将反应混合物加热至75℃搅拌反应16小时。待起始物料消耗完毕后,将反应混合物冷却至室温,用乙酸乙酯(30mL)稀释,后用水洗和盐水洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析色谱,并使用5%甲醇-CH2Cl2作为洗脱剂进行纯化,得到(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(化合物3525)为米白色固体(56mg,15%)。
1H NMR(400MHz,CD3OD)δ7.38–7.34(m,3H),7.22(dd,J=8.4Hz,5.2Hz,2H),7.09(d,J=1.6Hz,1H),6.98(t,J=8.8Hz,2H),6.94(d,J=8.8Hz,2H),6.80(dd,J=8.8Hz,2.4Hz,1H),4.20(t,J=6.8Hz,2H),3.63(br s,4H),2.87–2.76(m,2H),2.75–2.34(m,22H),2.18(s,3H),1.94(五重峰,J=6.8Hz,2H)。LCMS:m/z612.54[M+H]+。
通过此方法制备得到其他类似物:
化合物3526,4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(18%)。
1H NMR(300MHz,CD3OD)δ7.38–7.33(m,3H),7.12(d,J=8.7Hz,2H),7.09(d,J=2.1Hz,1H),6.94(d,J=9.0Hz,2H),6.86–6.77(m,3H),4.19(t,J=6.6Hz,2H),3.75(s,3H),3.63(br s,4H),2.80–2.71(m,2H),2.66–2.29(m,19H),2.28(s,3H),2.17(s,3H),1.98–1.87(m,2H)。LCMS:m/z 624.6[M+H]+。
化合物3527,(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)甲酮(9%)。
1H NMR(400MHz,CD3OD)δ7.37(d,J=8.4Hz,3H),7.10(d,J=2.0Hz,1H),6.94(d,J=8.4Hz,2H),6.82(dd,J=8.8Hz,2.0Hz,1H),6.74–6.66(m,3H),5.88(s,2H),4.23(t,J=6.8Hz,2H),3.66(br s,4H),2.95–2.40(m,21H),2.39(s,3H),2.18(s,3H),1.99–1.91(m,2H)。LCMS:m/z 638.52[M+H]+。
化合物3528,4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(25%)。
1H NMR(300MHz,CD3OD)δ7.40–7.33(m,3H),7.27(td,J=8.1Hz,6.0Hz,1H),7.09(d,J=2.4Hz,1H),7.07–6.85(m,5H),6.81(dd,J=8.7Hz,2.4Hz,1H),4.20(t,J=6.6Hz,2H),3.64(br s,4H),2.87–2.47(m,16H),2.47–2.34(m,8H),2.18(s,3H),1.94(五重峰,J=7.2Hz,2H)。LCMS:m/z 612.51[M+H]+。
化合物3529,4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮(26%)。
1H NMR(300MHz,CD3OD)δ7.40–7.33(m,3H),7.17(t,J=8.1Hz,1H),7.09(d,J=1.8Hz,1H),6.94(d,J=8.7Hz,2H),6.82–6.71(m,4H),4.19(t,J=7.2Hz,2H),3.76(s,3H),3.63(br s,4H),2.86–2.49(m,16H),2.43–2.32(m,5H),2.32(s,3H),2.18(s,3H),2.00–1.88(m,2H)。LCMS:m/z 624.52[M+H]+。
化合物3530,4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氧基)苯基)(4-苯乙基哌嗪-1-基)甲酮(12%)。
1H NMR(400MHz,CD3OD)δ7.37(d,J=8.4Hz,3H),7.29–7.14(m,5H),7.09(d,J=2.0Hz,1H),6.94(d,J=8.4Hz,2H),6.81(dd,J=9.2Hz,2.4Hz,1H),4.21(t,J=7.2Hz,2H),3.63(br s,4H),2.99–2.72(m,6H),2.72–2.46(m,13H),2.46–2.32(m,5H),2.18(s,3H),1.95(五重峰,J=6.8Hz 2H)。LCMS:m/z 594.52[M+H]+。
方案12.化合物3531–3536的一般合成
步骤1:(4-(3-甲氧基苯乙基)哌嗪-1-基)(4-硝基苯基)甲酮的制备
室温下,向1-(3-甲氧基苯乙基)哌嗪盐酸盐(500mg,2.99mmol)的DMF(10mL)的搅拌溶液中,加入HATU(2.27g,5.98mmol)、DIPEA(2.47mL,14.9mmol)和4-硝基苯甲酸(930mg,3.59mmol)。反应混合物在室温下搅拌反应16小时。待起始物料消耗完毕后,将反应混合物倒入冰水中,并用乙酸乙酯萃取。所得有机层用水洗和盐水洗,无水Na2SO4干燥,减压浓缩。所得粗产物经快速柱层析,并使用5%甲醇-CH2Cl2作为洗脱剂进行纯化,得到(4-(3-甲氧基苯乙基)哌嗪-1-基)(4-硝基苯基)甲酮为棕色固体(1.0g,90%)。LCMS:m/z 370.0[M+H]+。
通过此方法制备得到其他类似物:
(4-(4-氟苯乙基)哌嗪-1-基)(4-硝基苯基)甲酮(100%)
(4-(4-甲氧基苯乙基)哌嗪-1-基)(4-硝基苯基)甲酮(72%)
(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(4-硝基苯基)甲酮(27%)
(4-(3-氟苯乙基)哌嗪-1-基)(4-硝基苯基)甲酮(68%)
(4-硝基苯基)(4-苯乙基哌嗪-1-基)甲酮(75%)
步骤2:(4-氨基苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮的制备
室温下,向(4-(3-甲氧基苯乙基)哌嗪-1-基)(4-硝基苯基)甲酮(900mg,2.43mmol)的乙醇和水(1:1,各10mL)搅拌溶液中,加入Fe粉(300mg,12.16mmol)、和NH4Cl(325mg,6.08mmol)。将反应混合物加热至60℃搅拌反应3小时。待起始物料消耗完毕后,反应混合物通过硅藻土过滤,蒸去滤液中的乙醇。所得水层用乙酸乙酯萃取,合并的乙酸乙酯层用盐水洗,无水Na2SO4干燥,并减压浓缩得到粗产物。所得粗产物经快速柱层析,并使用6%甲醇-DCM作为洗脱剂进行纯化,得到(4-氨基苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮为棕色固体(300mg,41%)。LCMS:m/z 340.0[M+H]+。
通过此方法制备得到其他类似物:
(4-氨基苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(72%)
(4-氨基苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(42%)
(4-氨基苯基)(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)甲酮(43%)
(4-氨基苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(100%)
(4-氨基苯基)(4-苯乙基哌嗪-1-基)甲酮(74%)
步骤3:化合物3531,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮的制备
将(4-氨基苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(120mg,0.355mmol)和NaOtBu(78mg,0.82mmol)加至5-溴-2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚(100mg,0.273mmol)的1,4-二氧六环(3mL)搅拌溶液中。反应混合物通氩气10分钟进行脱气。然后加入Pd2(dba)3(17mg,0.019mmol)和2-二环己膦基-2'-(N,N-二甲胺)-联苯(16mg,0.041mmol),反应混合物继续通氩气10分钟进行脱气。将反应混合物加热至90℃搅拌反应16小时。待起始物料消耗完毕后,反应混合物用乙酸乙酯稀释,并通过硅藻土过滤。所得有机层用水洗和盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经制备TCL,并使用5%甲醇的CH2Cl2进行洗脱纯化,得到(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(4-氟苯乙基)哌嗪-1-基)甲酮(化合物3531)为淡黄色固体(20mg,21%)。
1H NMR(300MHz,d6-DMSO):δ8.11(br s,1H),7.33(d,J=8.4Hz,1H),7.26(dd,J=8.4Hz,5.7Hz,2H),7.20(d,J=8.4Hz,2H),7.15(br s,1H),7.09(t,J=9.0Hz,2H),6.92–6.83(m,3H),4.10(t,J=6.9Hz,2H),3.51(br s,4H),2.78–2.70(m,2H),2.64–2.15(m,22H),2.14(s,3H),1.85–1.76(m,2H)。LCMS:m/z 611.53[M+H]+。
通过此方法制备得到其他类似物:
化合物3532,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(42%)。
1H NMR(300MHz,d6-DMSO):δ8.12(br s,1H),7.33(d,J=8.4Hz,1H),7.20(d,J=8.4Hz,2H),7.17–7.10(m,3H),6.92–6.80(m,5H),4.10(t,J=6.6Hz,2H),3.71(s,3H),3.50(br s,4H),2.77–2.56(m,6H),2.48–2.19(m,18H),2.14(s,3H),1.86–1.75(m,2H)。LCMS:m/z 623.51[M+H]+。
化合物3533,(4-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)哌嗪-1-基)(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)甲酮(18%)。
1H NMR(400MHz,d6-DMSO):δ8.12(br s,1H),7.33(d,J=8.8Hz,1H),7.20(d,J=8.4Hz,2H),7.15(d,J=1.6Hz,1H),6.92–6.76(m,5H),6.67(br d,J=8.4Hz,1H),5.95(s,2H),4.09(t,J=6.8Hz,2H),3.50(br s,4H),2.69–2.61(m,2H),2.47–2.17(m,22H),2.14(s,3H),1.85–1.73(m,2H)。LCMS:m/z 637.47[M+H]+。
化合物3534,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(3-氟苯乙基)哌嗪-1-基)甲酮(11%)。
1H NMR(300MHz,d6-DMSO):δ8.11(br s,1H),7.37–7.26(m,2H),7.20(d,J=8.4Hz,2H),7.15(d,J=1.8Hz,1H),7.13–7.05(m,2H),7/00(td,J=8.7Hz,2.1Hz,1H),6.93–6.79(m,3H),4.09(t,J=6.9Hz,2H),3.50(br s,4H),2.81–2.72(m,2H),2.61–2.53(m,2H),2.48–2.29(m,15H),2.27–2.10(m,8H),1.84–1.72(m,2H)。LCMS:m/z 611.53[M+H]+。
化合物3535,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(3-甲氧基苯乙基)哌嗪-1-基)甲酮(30%)。
1H NMR(300MHz,CD3OD):δ7.90(br s,1H),7.35–7.12(m,5H),7.00–6.85(m,3H),6.83–6.72(m,3H),4.20(t,J=6.6Hz,2H),3.77(s,3H),3.71(br s,3H),3.15–2.89(m,4H),2.89–2.41(m,17H),2.37(s,3H),2.19(s,3H),2.03–1.89(m,2H)。LCMS:m/z 623.51[M+H]+。
化合物3536,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)氨基)苯基)(4-(4-甲氧基苯乙基)哌嗪-1-基)甲酮(10%)。
1H NMR(400MHz,d6-DMSO):δ8.11(br s,1H),7.41–7.09(m,9H),6.96–6.77(m,3H),4.09(br s,2H),3.50(br s,4H),2.78–2.70(m,2H),2.63–2.50(m,9H),2.39–2.09(m,16H),1.83–1.74(m,2H)。LCMS:m/z 593.55[M+H]+。
方案13.化合物3537–3540的一般合成
步骤1:2-(6-甲氧基萘-2-基)乙烷-1-醇的制备
0℃下,向LiAlH4(789mg,13.9mmol)的THF(100mL)混合物中加入2-(6-甲氧基萘-2-基)乙酸(3.00g,13.9mmol)。混合物在此温度下搅拌10分钟后,将反应体系慢慢升至室温,继续搅拌反应2小时。通过TCL检测,确定起始物料消耗完毕后,0℃下用乙酸乙酯(3mL)和饱和氯化铵溶液(20mL)淬灭反应,过滤,浓缩得到2-(6-甲氧基萘-2-基)乙烷-1-醇为白色固体(2.5g,95%)。
1H NMR(300MHz,d6-DMSO):δ7.89–7.80(m,3H),7.71(br s,1H),7.51–7.38(m,3H),4.67(t,J=5.1Hz,1H),3.70(q,J=6.9Hz,2H),2.89(t,J=6.9Hz,2H)。
通过此方法制备得到其他类似物:
2-(萘-2-基)乙烷-1-醇(77%)
2-(萘-1-基)乙烷-1-醇(86%)
2-(7-甲氧基萘-1-基)乙烷-1-醇(85%)
步骤2:2-(2-溴乙基)萘的制备
0℃下,向2-(萘-2-基)乙烷-1-醇(5.20g,30.2mmol)的DCM(100mL)溶液中,分批加入CBr4(10.99g,33.21mmol)和PPh3(8.70g,33.2mmol)。反应混合物搅拌10分钟后,将温度升至室温并搅拌反应2小时。通过TCL检测,确定起始物料消耗完毕后,向反应混合物中加入冰水,然后用DCM萃取。合并的有机层用水洗、然后盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析,并使用10-15%乙酸乙酯的石油醚作为洗脱剂进行纯化,得到2-(2-溴乙基)萘为黄色液体(5.2g,71%)。
通过此方法制备得到其他类似物:
2-(2-溴乙基)-6-甲氧基萘(79%)
1-(2-溴乙基)萘(73%)
1-(2-溴乙基)-7-甲氧基萘(78%)
步骤3:4-(2-(萘-2-基)乙基)哌嗪-1-羧酸叔丁酯的制备
室温下,向N-Boc哌嗪(4.94g,26.6mmol)的DMF(60mL)溶液中加入K2CO3(6.11g,44.3mmol)、2-(2-溴乙基)萘(5.2g,22.1mmol)和NaI(3.31g,22.1mmol)。将反应混合物加热至80℃搅拌反应12小时。通过TCL检测,确定起始物料消耗完毕后,向反应混合物中加入冰水,然后用乙酸乙酯萃取。合并有机层用水洗和盐水洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析,并使用40-50%乙酸乙酯的石油醚作为洗脱剂进行纯化,得到4-(2-(萘-2-基)乙基)哌嗪-1-羧酸叔丁酯为棕色胶状固体(7.0g,93%)。LCMS:m/z 341.35[M+H]+。
通过此方法制备得到其他类似物:
4-(2-(6-甲氧基萘-2-基)乙基)哌嗪-1-羧酸叔丁酯(58%)
4-(2-(萘-1-基)乙基)哌嗪-1-羧酸叔丁酯(46%)
4-(2-(7-甲氧基萘-1-基)乙基)哌嗪-1-羧酸叔丁酯(74%)
4-(2-(喹啉-6-基)乙基)哌嗪-1-羧酸叔丁酯
步骤4:1-(2-(萘-2-基)乙基)哌嗪盐酸盐的制备
0℃下,向4-(2-(萘-2-基)乙基)哌嗪-1-羧酸叔丁酯(7.00g,20.6mmol)的DCM(70mL)搅拌溶液中,加入1,4-二氧六环/HCl(10.3mL,~4M)。反应混合物搅拌10分钟后,将温度升至室温,然后反应混合物搅拌反应12小时。通过TCL检测,确定起始物料消耗完毕后,反应混合物减压浓缩,然后加入乙醚,将混合物过滤,得到1-(2-(萘-2-基)乙基)哌嗪盐酸盐为棕色固体(6.0g,89%)。LCMS:m/z 241.29[(M-HCl)+H]+。
通过此方法制备得到其他类似物:
1-(2-(6-甲氧基萘-2-基)乙基)哌嗪盐酸盐(91%)
1-(2-(萘-1-基)乙基)哌嗪盐酸盐(68%)
1-(2-(7-甲氧基萘-1-基)乙基)哌嗪盐酸盐(74%)
步骤5:(3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮的制备
0℃下,将HATU(511.0mg,1.344mmol)加至3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸(250mg,0.896mmol)和DIPEA(0.5mL)的DMF(5mL)搅拌溶液中。反应混合物在室温下搅拌30分钟。然后加入1-(2-(萘-2-基)乙基)哌嗪盐酸盐(273mg,0.986mmol),反应混合物在室温下搅拌反应过夜。反应进度通过TCL监测。待起始物料消耗完毕后,将反应混合物倒入冰水中。过滤,收集所析出沉淀,干燥。所得粗产物经快速柱层析,并使用5%甲醇的CH2Cl2作为洗脱剂进行纯化,得到(3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮为棕色固体(0.40g,86%)。LCMS:m/z 517.55[M+H]+。
通过此方法制备得到其他类似物:
(3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(6-甲氧基萘-2-基)乙基)哌嗪-1-基)甲酮(63%)
(3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-1-基)乙基)哌嗪-1-基)甲酮(63%)
(3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(7-甲氧基萘-1-基)乙基)哌嗪-1-基)甲酮(61%)
步骤6-1:(3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(6-甲氧基萘-2-基)乙基)哌嗪-1-基)甲酮的制备
0℃下,将NaH(102mg,2.56mmol)分批加至(3-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(6-甲氧基萘-2-基)乙基)哌嗪-1-基)甲酮(680mg,1.22mmol)的DMF(7mL)搅拌溶液中。然后将混合物升至室温,搅拌30分钟。0℃下向反应混合物中滴加溴氯丙烷(1.20mL,6.49mmol),然后在室温下搅拌反应3小时。待起始物料消耗完毕后,向反应混合物中加入冰水,然后用乙酸乙酯萃取。有机层用盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析,并使用乙酸乙酯作为洗脱剂进行纯化,得到(3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(6-甲氧基萘-2-基)乙基)哌嗪-1-基)甲酮为棕色胶状固体(250mg,42%)。LCMS:m/z 608.3[M+H]+。
通过此方法制备得到其他类似物:
(3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮(98%)
(3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-1-基)乙基)哌嗪-1-基)甲酮(66%)
(3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(7-甲氧基萘-1-基)乙基)哌嗪-1-基)甲酮(44%)
步骤6-2:化合物3537,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮的制备
室温下,向(3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮(150mg,0.284mmol)的乙腈(5mL)搅拌溶液中,加入碘化钠(85.09mg,0.5680mmol)和碳酸钠(90.31mg,0.8520mmol),随后加入N-甲基哌嗪(71.10mg,0.7100mmol)。然后将反应混合物加热至75℃搅拌反应16小时。待起始物料消耗完毕后,将反应混合物冷却至室温,用乙酸乙酯(40mL)稀释。混合物用水洗和盐水溶液洗,无水Na2SO4干燥,减压浓缩。所得粗产物经快速柱层析,并使用5%甲醇-CH2Cl2作为洗脱剂进行纯化,得到(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮(化合物3537)为淡黄色固体(35mg,11%)。
1H NMR(300MHz,CD3OD):δ8.05(br d,J=8.1Hz,1H),7.88(dd,J=7.5Hz,1.5Hz,1H),7.75(br d,J=8.1Hz,1H),7.57–7.32(m,6H),7.34–7.18(m,3H),7.09(br s,1H),6.90(dd,J=8.1Hz,1.5Hz,1H),4.08(s,2H),3.93(t,J=6.9Hz,2H),3.73(br s,2H),3.28–3.20(m,4H),2.68–2.10(m,25H),1.77(五重峰,J=6.9Hz,2H)。LCMS:m/z 642.54[M+H]+。
通过此方法制备得到其他类似物:
化合物3538,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(6-甲氧基萘-2-基)乙基)哌嗪-1-基)甲酮(23%)。
1H NMR(300MHz,d6-DMSO):δ7.84(d,J=8.7Hz,1H),7.70(br d,J=7.5Hz,1H),7.38–7.14(m,10H),6.90(br d,J=8.4Hz,1H),4.07–3.98(m,4H),3.89(s,3H),3.61(br s,2H),3.33(br s,2H),3.21–3.12(m,2H),2.65–2.11(m,25H),1.76–1.64(m,2H)。LCMS:m/z 672.57[M+H]+。
化合物3539,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-1-基)乙基)哌嗪-1-基)甲酮(22%)。
1H NMR(300MHz,CD3OD):δ8.05(br d,J=8.1Hz,1H),7.88(br d,J=7.2Hz,1H),7.76(br d,J=7.8Hz,1H),7.58–7.33(m,6H),7.26–7.15(m,3H),7.09(br s,1H),6.91(br d,J=8.1Hz,1H),4.09(s,2H),3.93(t,J=6.6Hz,2H),3.74(br s,2H),3.25–3.19(m,4H),2.59–2.15(m,19H),2.14(s,3H),2.11(s,3H),1.78(五重峰,J=6.9Hz,2H)。LCMS:m/z 642.54[M+H]+。
化合物3540,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(7-甲氧基萘-1-基)乙基)哌嗪-1-基)甲酮(5%)。
1H NMR(300MHz,d6-DMSO):δ7.76–7.68(m,2H),7.63(br s,1H),7.37–7.31(m,3H),7.29–7.23(m,3H),7.19–7.10(m,3H),6.91(br d,J=9.6Hz,1H),4.10–3.98(m,4H),3.84(s,3H),3.58(br s,4H),2.88–2.79(m,2H),2.63–2.11(m,25H),1.81–1.68(m,2H)。LCMS:m/z 672.53[M+H]+。
方案14.化合物3542–3545的一般合成
步骤1:(4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮的制备
0℃下,将HATU(680mg,1.79mmol)加至4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸(250mg,0.896mmol)、和DIPEA(0.45mL,2.68mmol)的DMF(5mL)搅拌溶液中。然后加入1-(2-(萘-2-基)乙基)哌嗪盐酸盐(276mg,0.997mmol),反应混合物在室温下搅拌反应16小时。反应进度通过TCL监测。待起始物料消耗完毕后,将反应混合物倒入冰水中,用乙酸乙酯萃取。合并的有机层用盐水洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析,并使用5%甲醇的CH2Cl2作为洗脱剂进行纯化,得到(4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮为棕色固体(400mg,86%)。LCMS:m/z 502.24[M+H]+。
通过此方法制备得到其他类似物:
(4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(6-甲氧基萘-2-基)乙基)哌嗪-1-基)甲酮(92%)
(4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-1-基)乙基)哌嗪-1-基)甲酮(90%)
(4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(7-甲氧基萘-1-基)乙基)哌嗪-1-基)甲酮(90%)
步骤2-1:(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮的制备
0℃下,将NaH(15mg,0.63mmol)分批加至(4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮(150mg,0.290mmol)的DMF(6mL)搅拌溶液中。将混合物升至室温,搅拌30分钟。0℃下向反应体系中滴加溴氯丙烷(0.15mL,1.5mmol)。反应混合物在室温下搅拌反应3小时。待起始物料消耗完毕后,向反应混合物中加入冰水,然后用乙酸乙酯萃取。所得有机层用盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析,并使用乙酸乙酯作为洗脱剂进行纯化,得到(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮为黄色液体(170mg,98%)。LCMS:m/z 578.43[M+H]+。
通过此方法制备得到其他类似物:
(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(6-甲氧基萘-2-基)乙基)哌嗪-1-基)甲酮(42%)
(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-1-基)乙基)哌嗪-1-基)甲酮(36%)
(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(7-甲氧基萘-1-基)乙基)哌嗪-1-基)甲酮(44%)
步骤6-2:化合物3542,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮的制备
室温下,向(4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮(170mg,0.284mmol)的乙腈(5mL)搅拌溶液中,加入碘化钠(88.09mg,0.58mmol)和碳酸钠(155mg,1.47mmol),随后加入N-甲基哌嗪(117mg,1.17mmol)。将反应混合物加热至75℃搅拌反应16小时。待起始物料消耗完毕后,将反应混合物冷却至室温,用乙酸乙酯(40mL)稀释,然后用水洗,随后用盐水洗。所得有机层用无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析,并使用5%甲醇-DCM作为洗脱剂进行纯化,得到(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-2-基)乙基)哌嗪-1-基)甲酮(化合物3524)为黄色粘性液体(25mg,14%)。
1H NMR(400MHz,CD3OD):δ8.07(br d,J=8.8Hz,1H),7.85(br d,J=7.6Hz,1H),7.72(dd,J=7.2Hz,2.0Hz,1H),7.53–7.44(m,2H),7.41–7.31(m,6H),7.24(br s,1H),7.21(d,J=8.4Hz,1H),6.92(dd,J=8.4Hz,1.6Hz,1H),4.13(t,J=6.8Hz,2H),4.07(s,2H),3.82(br s,2H),3.59(br s,2H),3.32–3.25(m,2H),2.77–2.35(m,14H),2.34(s,3H),2.31(t,J=6.8Hz,2H),2.26(s,3H),2.18(s,3H),1.89(五重峰,J=6.8Hz,2H)。LCMS:m/z642.57[M+H]+。
通过此方法制备得到其他类似物:
化合物3543,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(6-甲氧基萘-2-基)乙基)哌嗪-1-基)甲酮(12%)。
1H NMR(300MHz,d6-DMSO):δ7.75–7.70(m,2H),7.63(br s,1H),7.34(br d,J=9.9Hz,1H),7.31–7.23(m,7H),7.12(dd,J=8.7Hz,2.1Hz,1H),6.92(br d,J=9.6Hz,1H),4.07(t,J=6.6Hz,2H),4.02(br s,2H),3.85(s,3H),3.50(br s,4H),2.89–2.81(m,2H),2.64–2.58(m,2H),2.51–2.13(m,23H),1.75(五重峰,J=6.6Hz,2H)。LCMS:m/z 672.0[M+H]+。
化合物3544,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(萘-1-基)乙基)哌嗪-1-基)甲酮(33%)。
1H NMR(300MHz,d6-DMSO):δ8.04(br d,J=8.4Hz,1H),7.91(dd,J=7.5Hz,1.5Hz,1H),7.77(br d,6.9Hz,1H),7.58–7.45(m,2H),7.44–7.37(m,2H),7.31–7.23(m,6H),6.92(br d,J=9.6Hz,1H),4.07(t,J=7.2Hz,2H),4.02(br s,2H),3.55(br s,4H),3.24–3.18(m,2H),2.67–2.60(m,2H),2.47–2.12(m,23H),1.75(五重峰,J=6.9Hz,2H)。LCMS:m/z 642.1[M+H]+。
化合物3545,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(7-甲氧基萘-1-基)乙基)哌嗪-1-基)甲酮(33%)。
1H NMR(300MHz,d6-DMSO):δ7.84(d,J=9.2Hz,1H),7.70(br d,J=8.4Hz,1H),7.36–7.23(m,7H),7.20–7.15(m,3H),6.90(dd,J=8.4Hz,1.6Hz,1H),4.06–3.99(m,4H),3.61(br s,4H),3.18–3.11(m,2H),2.63–2.11(m,25H),1.71(五重峰,J=7.2Hz,2H)。LCMS:m/z 672.1[M+H]+。
方案15.化合物3541和3546的一般合成
步骤1:2-(喹啉-6-基)乙酸甲酯的制备
向2-(喹啉-6-基)乙酸(2.0g,11mmol)的甲醇(40mL)溶液中加入浓硫酸(0.2mL)。反应混合物在室温下搅拌反应6小时。然后将反应混合物倒入水中,所得溶液用饱和NaHCO3溶液中和,然后用乙酸乙酯萃取。所得有机层用水洗和盐水洗,Na2SO4干燥,真空减压浓缩得到2-(喹啉-6-基)乙酸甲酯(1.8g,85%)。LCMS:m/z 202.25[M+H]+。
步骤2:2-(喹啉-6-基)乙烷-1-醇的制备
0℃下,向LiAlH4(680mg,17.9mmol)的THF(50mL)混合物中加入2-(喹啉-6-基)乙酸甲酯(1.8g,8.9mmol)。将混合物在此温度下搅拌10分钟,然后将反应体系慢慢升至室温,继续搅拌反应4小时。通过TCL检测,确定起始物料消耗完毕后,0℃下用乙酸乙酯(3mL)和饱和氯化铵溶液(20mL)淬灭反应,过滤,减压浓缩得到2-(喹啉-6-基)乙烷-1-醇(1.3g,84%)。LCMS:m/z 174.28[M+H]+。
步骤3:2-(喹啉-6-基)乙基甲磺酸酯的制备
0℃下,向2-(喹啉-6-基)乙烷-1-醇(1.3g,7.5mmol)的二氯甲烷(25mL)溶液中加入三乙胺(5.2mL,38mmol)和甲磺酰氯(1.2mL,15mmol)。反应混合物在此温度下搅拌反应3小时。通过TCL检测,确定起始物料消耗完毕后,将反应混合物倒入水中,然后用乙酸乙酯萃取。所得有机萃取液用水洗和盐水洗,Na2SO4干燥,蒸发浓缩得到2-(喹啉-6-基)乙基甲磺酸酯(1.7g,粗品),其可直接使用无需进一步纯化。
步骤4:4-(2-(喹啉-6-基)乙基)哌嗪-1-羧酸叔丁酯的制备
室温下,向N-Boc哌嗪(1.3g,7.2mmol)的DMF(60mL)溶液中加入K2CO3(1.49g,10.8mmol)和2-(喹啉-6-基)乙基甲磺酸酯(0.9g,3.6mmol)。将反应混合物加热至80℃搅拌反应12小时。通过TCL检测,确定起始物料消耗完毕后,向反应混合物中加入冰水,然后用乙酸乙酯萃取。合并的有机层用水洗和盐水洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析,并使用40-50%乙酸乙酯的石油醚作为洗脱剂进行纯化,得到4-(2-(喹啉-6-基)乙基)哌嗪-1-羧酸叔丁酯(0.8g,66%)。LCMS:m/z 342.05[M+H]+。
步骤5:6-(2-(哌嗪-1-基)乙基)喹啉盐酸盐的制备
0℃下,向4-(2-(喹啉-6-基)乙基)哌嗪-1-羧酸叔丁酯(800mg,2.3mmol)的DCM(50mL)搅拌溶液中,加入1,4-二氧六环/HCl(25mL,~4M)。反应混合物搅拌10分钟后,将温度升至室温,并搅拌反应12小时。通过TCL检测,确定起始物料消耗完毕后,将反应混合物浓缩,加入乙醚,过滤,得到6-(2-(哌嗪-1-基)乙基)喹啉盐酸盐(0.6g,粗品)。LCMS:m/z 242.2[(M-HCl)+H]+。
步骤6-1:4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸甲酯的制备
0℃下,将NaH(400mg,16.7mmol)分批加至4-((2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸甲酯(830mg,2.8mmol)的DMF(10mL)搅拌溶液中。将混合物升至室温搅拌反应30分钟。0℃下,向反应体系中滴加溴氯丙烷(0.6g,3.8mmol),然后反应混合物在此温度下搅拌反应2小时。待起始物料消耗完毕后,向反应混合物中加入冰水,然后用乙酸乙酯萃取。所得有机层用盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析纯化,得到4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸甲酯(380mg,36%)。
1H NMR(400MHz,CDCl3):δ7.93(d,J=8.4Hz,2H),7.30–7.27(m,3H),7.20(d,J=8.0Hz,1H),6.94(dd,J=8.0Hz,1.6Hz,1H),4.22(t,J=6.8Hz,2H),4.18(s,2H),3.89(s,2H),3.50(t,J=6.0Hz,2H),2.35(s,3H),2.22–2.16(m,5H)。LCMS:m/z 370.1[M+H]+。
通过此方法制备得到其他类似物:
3-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸甲酯(32%)。
步骤6-2:4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯甲酸甲酯的制备
室温下,向4-((1-(3-氯丙基)-2,3-二甲基-1H-吲哚-5-基)甲基)苯甲酸甲酯(380mg,1.0mmol)的乙腈(12mL)搅拌溶液中,加入碘化钠(380mg,2.5mmol)和碳酸钠(270mg,2.5mmol),随后加入N-甲基哌嗪(250mg,2.5mmol)。将反应混合物升至80℃搅拌反应12小时。待起始物料消耗完毕后,将反应混合物冷却至室温,用乙酸乙酯稀释,然后用水洗和盐水溶液洗,无水Na2SO4干燥,减压浓缩得到粗产物。所得粗产物经快速柱层析色谱纯化,得到4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯甲酸甲酯(310mg,67%)。
1H NMR(400MHz,CDCl3):δ7.93(d,J=8.4Hz,2H),7.29–7.26(m,3H),7.18(d,J=8.4Hz,1H),6.93(dd,J=8.4Hz,1.2Hz,1H),4.12–4.07(m,4H),3.89(s,3H),2.82–2.31(m,16H),2.20(s,3H),1.92–1.85(m,2H)。LCMS:m/z 434.3[M+H]+。
通过此方法制备得到其他类似物:
3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯甲酸甲酯(67%)。
步骤7:4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯甲酸的制备
室温下,向4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯甲酸甲酯(200mg,0.46mmol)的THF:H2O:MeOH(4:1:1,5mL)溶液中加入LiOH.H2O(38mg,0.91mmol)。反应混合物搅拌反应12小时。待起始物料消耗完毕后,将反应混合物浓缩,然后加入乙酸乙酯和水,使其分层。收集水层,并在0℃下用饱和柠檬酸溶液酸化。过滤,收集所析出的沉淀,真空干燥,得到4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯甲酸(160mg,84%)。
通过此方法制备得到其他类似物:
3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯甲酸(75%)。LCMS:m/z 420.3[M+H]+。
步骤8:化合物3541,(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(喹啉-6-基)乙基)哌嗪-1-基)甲酮的制备
向3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯甲酸(90mg,0.21mmol)和6-(2-(哌嗪-1-基)乙基)喹啉盐酸盐(175mg,0.63mmol)的吡啶(3mL)溶液中,加入EDC.HCl(230mg,1.2mmol)。混合物在80℃下搅拌反应1小时。然后向反应混合物中加入乙酸乙酯和水,使其分层。所得有机层用水洗和盐水洗,然后干燥,减压浓缩。所得残留物经制备HPLC纯化得到(3-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(喹啉-6-基)乙基)哌嗪-1-基)甲酮(化合物3541)(8mg,6%)。
1H NMR(400MHz,DMSO-d6):δ8.83(m,1H),8.28(br d,J=7.6Hz,1H),7.94(d,J=8.4Hz,1H),7.79(br s,1H),7.68–7.63(m,1H),7.55–7.49(m,1H),7.38–7.32(m,2H),7.28–7.22(m,2H),7.19–7.13(m,2H),6.91(br d,J=8.8Hz,1H),4.09-4.00(m,4H),3.41–3.19(m,4H),2.97–2.89(m,2H),2.64–2.59(m,2H),2.39–2.08(m,23H),1.77-1.69(m,2H)。LCMS:m/z 643.4[M+H]+。
通过此方法制备得到其他类似物:
化合物3546,(4-((2,3-二甲基-1-(3-(4-甲基哌嗪-1-基)丙基)-1H-吲哚-5-基)甲基)苯基)(4-(2-(喹啉-6-基)乙基)哌嗪-1-基)甲酮(2%)。
1H NMR(400MHz,DMSO-d6):δ8.83(dd,J=4.4Hz,2.0Hz,1H),8.27(br d,J=7.6Hz,1H),7.93(d,J=8.4Hz,1H),7.78(br s,1H),7.65(dd,J=8.8Hz,2.0Hz,1H),7.49(dd,J=8.4Hz,4.0Hz,1H),7.31–7.24(m,6H),6.91(br d,J=9.2Hz,1H),4.11–4.02(m,4H),3.58(br s,4H),2.97–2.91(m,2H),2.68–2.61(m,2H),2.50–2.11(m,20H),1.85(s,3H),1.79–1.72(m,2H)。LCMS:m/z 643.4[M+H]+。
作为单一疗法的抗原肌球蛋白化合物的活性
本发明化合物的抗增殖活性
在玻片模型中已识别在原肌球蛋白Tpm3.1上的结合位点,从而产生一系列本发明化合物作为原肌球蛋白抑制剂。在肿瘤细胞中,抑制Tpm3.1将导致肌动蛋白细胞骨架的破坏和最终细胞死亡。
对化合物3501-3540和3542-3545抑制肿瘤细胞典型如神经母细胞瘤、黑色素瘤、前列腺癌、结直肠癌、非小细胞肺癌、和三阴性乳腺癌细胞增殖的效能进行评价。简言之,将通过对每个受试细胞株的细胞生长试验来计算得到预先确定的细胞数量,即将每个受试的细胞株接种于其各自的培养基中(采用ATCC培养参数),并在37℃、5%CO2下,于96-孔培养板中,培育24小时。一旦附着,便将每个细胞株暴露于每个各自类似物增加的浓度中(对于表1中化合物,浓度分别为0.03、0.3、3和30μM;对于表2中化合物,浓度分别为0.1、0.3、1、3、10和30μM),继续培育72小时,然后进一步暴露于细胞-滴定度发光试剂(100μL/孔)中,继续培育30分钟,从而评价细胞存活率。冷发光是采用EnVision多标记酶标仪进行捕捉的,且每个类似物浓度的数据与未经处理的对照进行对比。对于化合物3501、3503、3505–08、3512–18、3520、3522–24和3531–36,制作对照与浓度百分比的半对数曲线图,IC50是采用线性回归分析来确定的。对于化合物3502、3504、3509–11、3519、3521、3525–30、3537–40和3542–45,细胞存活率是通过对照(单独媒介物)与剂量-响应曲线进行归一化,且半数最大有效浓度(EC50)值采用Graph Pad Prism6(非线性回归反曲剂量–响应变量斜率)来确定。
表1.本发明化合物对一系列体癌细胞的抗增殖活性
表2.本发明化合物对一系列体癌细胞的抗增殖活性
针对化合物3507对代表性细胞株黑色素瘤、前列腺癌、白血病和神经母细胞瘤的抗增殖活性作出进一步评价。将细胞株暴露于化合物3507增加的浓度中72小时后的细胞存活率是采用MTS存活率试验来进行测定。细胞存活率是通过对照(单独媒介物)与剂量响应曲线进行归一化,且相对抑制浓度(IC50)值是通过Graph Pad Prism 6来确定。
表3.化合物3507对一系列体癌细胞的抗增殖活性
本发明化合物对肌动蛋白细胞骨架的影响
化合物3504、3507和3516破坏整个肌动蛋白细胞骨架的效能(图1)和特定靶向作用于包含Tpm3.1的肌动蛋白微丝(图2),是通过体外微丝破坏试验来进行评价。
简言之,将SK-N-SH神经母细胞瘤细胞接种于1800细胞/孔的384Perkin Elmer高含量成像“视图”孔板中,并在处理前培育24小时。然后,细胞由0-40μM的受试化合物处理(1:2于10点剂量响应连续稀释)。处理24h后,细胞被4%多聚甲醛(PBS)固定,并用Triton-X-100进行渗透化和用488-Atto-Phallodin染色,并通过DAPI来可视观察肌动蛋白微丝束和细胞核,或使用γ9d(羊多克隆,1:100),随后使用488-共轭第二抗体(1:1000)和DAPI来分别可视观察包含Tpm3.1的微丝束和细胞核。单一平面图像是通过使用20x目标的Perkin Elmer Opera共聚焦显微镜来得到。每一条件下视图的十二领域均被成像。而后将成像导出,并且组织和细胞内激动蛋白微丝数量的变化,是通过CSIRO开发的线性特征检测算法来进行数量测定。所述算法发现“山脊线”或“峰”是在局部像素强度的细胞成像中。所述这些“山脊线”与肌动蛋白微丝束相对应,以此来定量每个细胞中微丝的数量。
数据表明,化合物3504、3507和3516以剂量依赖方式,并均可破坏整个肌动蛋白细胞骨架和包含Tpm3.1的肌动蛋白微丝。
为了证明本发明化合物可损害Tpm3.1的功能,化合物3507对Tpm3.1调节的激动蛋白微丝解聚作用的影响,是通过良好表征的基于芘的肌动蛋白微丝解聚作用试验(Broschat,1990;Kostyukova和Hitchcock,2004)来进行评价。所述试验小结和基本原理如下所示:为了促进解聚作用,芘标记的肌动蛋白微丝是采用下述所指出终点的临界浓度来进行稀释(0.5μM,由Pollard等人规定,1986)。荧光下降是通过随着时间推移,肌动蛋白单体的分裂来进行测定的。从而证实,在Tpm3.1的存在下,肌动蛋白解聚作用的比率在显著减少(Bonello 2013)。因此,任何化合物作用并影响Tpm3.1的功能,将使Tpm3.1在肌动蛋白解聚作用上的保护作用无效。
对于所有单独的F-肌动蛋白解聚作用试验,Tpm3.1的人同源物包衣的F-肌动蛋白被作为比较对照。简言之,在稀释微丝之前,Tpm3.1用F-肌动蛋白预孵化20分钟,使Tpm3.1聚合物适当组装。不出所料,在饱和量的Tpm3.1存在下,F-肌动蛋白解聚作用的初始速率(V0)显著慢于包含Tpm3.1的肌动蛋白微丝(图3A和C,p<0.0001)。
而后,单独的F-肌动蛋白和用Tpm3.1包衣的F-肌动蛋白的解聚作用,是在受试化合物存在下,通过比较解聚作用初始速率来测定的。如上所述,在被加至肌动蛋白微丝中之前,Tpm3.1在50μM化合物3507中预孵化。在化合物3507存在下,Tpm3.1聚合和保护肌动蛋白的能力被损害,且解聚速率与F-肌动蛋白无明显差异(图3B和D)。所述这些数据表明,化合物3507可以作用并损害Tpm3.1的功能。
本发明化合物对细胞因子释放的影响
对化合物3507、3520、3534和3538抑制细胞因子TNF-α、IFN-γ、IL-6、IL-21、IL-17A和IL-23释放的效能在体外进行评价(表4和5)。简言之,人外周血单核细胞(PBMCs)从人外周血通过Histopaque密度梯度离心法分离得到。将新鲜分离的PBMCs接种于50,000细胞株/孔的96-孔半区板中。PBMCs按剂量给予受试化合物(以10μM、1μM和0.1μM),然后在37℃、5%CO2下孵育2小时。为了刺激细胞因子IFN-γ、IL-21、IL-17A和IL-23的释放,将PBMCs用50ng/mL的佛波醇12-十四酸酯13-乙酸酯(PMA)和1μg/mL的离子霉素进行处理,从而刺激TNF-α和IL-6的释放,将PBMCs用100ng/mL来自革兰氏阴性菌的脂多糖(LPS)进行处理。然后将PBMCs在37℃、5%CO2下,进一步孵育6小时,收集细胞上清液,采用均相时间分辨荧光法(HTRF)并根据制造商说明书进行分析。细胞因子从所述PBMCs释放是采用Perkin Elmer ENVISION 2104酶标仪分别设定在615nm和665nm进行捕捉的。细胞毒性的分析是在相似条件下使用按剂量给予相同受试化合物的100,000PBMCs的96-孔板,在2小时的时间点有或没有PMA和离子霉素进行刺激,显示的任何发生的小细胞消亡,均足以说明在每六个实验中观察到的对细胞因子释放的抑制。
表4.本发明化合物对一系列细胞因子的抑制活性
表5.本发明化合物对一系列细胞因子的抑制活性
化合物3507的耐受性和体内药效
化合物3507的体内药效是通过神经母细胞瘤异种移植模型CHLA20进行评价的。CHLA20肿瘤是通过在裸鼠右侧皮下注射1.0x107肿瘤细胞,从而在无胸腺裸鼠中建立。当肿瘤体积达到~200-400mm3时,对动物开始给药。将动物(~n=5+)随机分为治疗组和对照组。化合物3507按剂量每天以150mg/kg通过腹腔内注射(IP)30%w/v磺丁基-β-环糊精(Captisol,包含环糊精的制剂)对动物进行给药。通过18天治疗后,发现化合物3507相较媒介物对照,具有良好的耐受性,并能显著减慢肿瘤生长(图4)。
还对化合物3507在人黑色素瘤(A375)异种移植模型的体内药效进行了评价。A375肿瘤是通过在小鼠右侧区域皮下注射约五百万肿瘤细胞,从而在雌性Foxn-1nu/nu无胸腺裸鼠中建立。当肿瘤体积达到130-150mm3时,将所述动物随机分成四组,(n=8或12只动物/组),以便所有组别的平均肿瘤体积相同。组别1给予媒介物(30%w/v Dexolve-7的无菌水),每周静脉注射两次,组别2按剂量60mg/kg给予3507/Dexolve-7,每周静脉注射两次。肿瘤和体重一周测定两或三次。此外,在整个研究期间,每天监测小鼠的临床状况。在整个研究期间,采用化合物3507处理后的动物的体重与对照组相当,表明化合物3507有良好的耐受性(图5A)。与神经母细胞瘤的研究一致,采用化合物3507治疗14天后,发现黑色素瘤肿瘤生长显著减少了~60%,相较媒介物对照(图5B)。
参考文献
Broschat,K.O.(1990).Tropomyosin prevents depolymerisation of actin filaments from the pointed end.J Biol Chem 265,21323-21329.
Kostyukova,A.S.,and Hitchcock-DeGregori,S.E.(2004).Effect of the structure of the N terminus of tropomyosin on tropomodulin function.J Biol Chem 279,5066-5071.
Pollard,T.D.(1986).Rate constants for the reactions of ATP-and ADP-actin with the ends of actin filaments.J Cell Biol 103,2747-2754.
Bonello,T.B(2013).Characterising the impact of tropomyosin targeting compounds in the actin cytoskeleton.Ph.D thesis,School of Medical Sciences,University of New South Wales,Australia
Vindin,H.,Bischof,L.,Gunning,P.&Stehn,J.Validation of an algorithm to quantify changes in actin cytoskeletal organization.J Biomol Screen 19,354-368(2014).
须知,本发明说明书公开和定义的内容,可延伸至所有可供选择的两个或多个本发明上下文或附图所述的个体特征的组合。所有这些不同的组合构成本发明各种可供选择和替代的方面。
- 还没有人留言评论。精彩留言会获得点赞!