2023/12/6最新更新专利技术
-
 .本发明属于生物,具体涉及冷冻保存骨组织的方法。.虽然采用人造骨骼能够快速解决骨组织缺损及防止因骨癌等因素回植造成的复发等问题,但人造骨骼材料的价格远超患者经济承受范围,同时无法避免患者因接受人造骨骼移植后出现“物非原主”导致的心理焦虑和不可预期的心理压力。从提高患者康复效果的角度...
.本发明属于生物,具体涉及冷冻保存骨组织的方法。.虽然采用人造骨骼能够快速解决骨组织缺损及防止因骨癌等因素回植造成的复发等问题,但人造骨骼材料的价格远超患者经济承受范围,同时无法避免患者因接受人造骨骼移植后出现“物非原主”导致的心理焦虑和不可预期的心理压力。从提高患者康复效果的角度... -
 .本发明涉及地板加工,具体为一种地板加工开槽装置及其开槽方法。.地板是一种地面装饰材料,根据材质,它可以分为实木地板、复合地板、竹地板、玻璃地板、金属地板等类型。根据型制,它可以分为活动地板、网络地板、普通地板等类型。地板大致可分为六大类:实木地板,实木复合地板,负离子木地板,自然...
.本发明涉及地板加工,具体为一种地板加工开槽装置及其开槽方法。.地板是一种地面装饰材料,根据材质,它可以分为实木地板、复合地板、竹地板、玻璃地板、金属地板等类型。根据型制,它可以分为活动地板、网络地板、普通地板等类型。地板大致可分为六大类:实木地板,实木复合地板,负离子木地板,自然... -
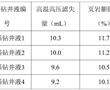 .本发明涉及页岩抑制剂,尤其涉及一种沥青类页岩抑制剂及其制备方法和应用。.页岩抑制剂俗称防塌剂,是指主要用来抑制页岩中所含黏土矿物的水化、膨胀、分解作用,以防止井塌的处理剂。常用的页岩抑制剂有石膏、硅酸盐、石灰、各种钾盐、铵盐、各种沥青制品以及高聚物的钾、铵、钙盐等。.目前,钻井液...
.本发明涉及页岩抑制剂,尤其涉及一种沥青类页岩抑制剂及其制备方法和应用。.页岩抑制剂俗称防塌剂,是指主要用来抑制页岩中所含黏土矿物的水化、膨胀、分解作用,以防止井塌的处理剂。常用的页岩抑制剂有石膏、硅酸盐、石灰、各种钾盐、铵盐、各种沥青制品以及高聚物的钾、铵、钙盐等。.目前,钻井液... -
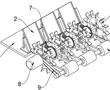 .本发明涉及水稻插秧领域,具体为一种水稻地膜覆盖插秧装置及插秧方法。.插秧,指将秧苗栽插于水田中,或指把水稻秧苗从秧田移植到稻田里。育种的时候水稻比较密集,不利于生长,经过人工移植或机器移植,让水稻有更大的生长空间,插秧时通常会用插秧机进行操作,插秧机是将稻苗植入稻田中的一种农业机械。进行...
.本发明涉及水稻插秧领域,具体为一种水稻地膜覆盖插秧装置及插秧方法。.插秧,指将秧苗栽插于水田中,或指把水稻秧苗从秧田移植到稻田里。育种的时候水稻比较密集,不利于生长,经过人工移植或机器移植,让水稻有更大的生长空间,插秧时通常会用插秧机进行操作,插秧机是将稻苗植入稻田中的一种农业机械。进行... -
.本发明涉及防腐剂领域,具体涉及一种具有高效抗氧化抑菌效果的组合物及其应用。.从农场到餐桌的整个环节中,食品均易受到微生物的侵染而发生腐败。全球每年约有.亿吨的食物因食品腐败被浪费,约占总生产产出的三分之一。化学防腐剂有低毒性和引起细菌耐药性的风险,故寻找安全的天然食品原料作为抑菌抗氧化进...
-
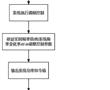 .本发明涉及一种分布式光储微电网调频控制方法,属于可再生能源发电领域。.随着配电台区、微电网中分布式光伏渗透率不断升高,配电台区、微电网的调频能力、调频容量愈发不足。传统仅依赖于微电网系统内的储能系统进行调频控制,需要较高的投资维护成本,且未有效利用光伏发电的调节潜力。因此亟需合适的光伏发...
.本发明涉及一种分布式光储微电网调频控制方法,属于可再生能源发电领域。.随着配电台区、微电网中分布式光伏渗透率不断升高,配电台区、微电网的调频能力、调频容量愈发不足。传统仅依赖于微电网系统内的储能系统进行调频控制,需要较高的投资维护成本,且未有效利用光伏发电的调节潜力。因此亟需合适的光伏发... -
 .本发明属于医疗康复,涉及一种脑-器械交互和脑-器官交互模型的迭代方法及相关装置。.脑器交互(brain-apparatuscommunications,bac)是近年涌现出的一个新观念,它强调整合大脑、身体和外部环境/器械的相互作用。bac一般分为体内脑器交互(bac-:“大脑...
.本发明属于医疗康复,涉及一种脑-器械交互和脑-器官交互模型的迭代方法及相关装置。.脑器交互(brain-apparatuscommunications,bac)是近年涌现出的一个新观念,它强调整合大脑、身体和外部环境/器械的相互作用。bac一般分为体内脑器交互(bac-:“大脑... -
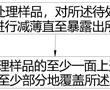 tem样品及其制备方法.本发明涉及半导体集成电路制造,特别涉及一种tem样品及其制备方法。.透射电子显微镜(transmissionelectronmicroscope,简称tem),是利用高能电子束穿透tem样品发生散射、吸收、干涉及衍射等现象,在成像平面形成衬度,从...
tem样品及其制备方法.本发明涉及半导体集成电路制造,特别涉及一种tem样品及其制备方法。.透射电子显微镜(transmissionelectronmicroscope,简称tem),是利用高能电子束穿透tem样品发生散射、吸收、干涉及衍射等现象,在成像平面形成衬度,从... -
 显示面板及显示装置.本发明涉及显示,尤其涉及一种显示面板及显示装置。.微型发光二极管(例如microled)显示面板因其发光效率高、结构紧凑、可靠性优等优良特性,广泛应用于各类显示领域中。.目前,为实现该类显示面板的触控功能,一般是采用外挂的方式在显示基板外侧设置...
显示面板及显示装置.本发明涉及显示,尤其涉及一种显示面板及显示装置。.微型发光二极管(例如microled)显示面板因其发光效率高、结构紧凑、可靠性优等优良特性,广泛应用于各类显示领域中。.目前,为实现该类显示面板的触控功能,一般是采用外挂的方式在显示基板外侧设置... -
 基于单张rgb图像实现不同人体重建模型的融合方法.本发明涉及计算机视觉处理领域,尤其涉及一种基于单张rgb图像实现不同人体重建模型的融合方法。.人体三维重建是三维重建领域中的的重要细分领域。低成本、便捷的获取人体三维模型有广泛的应用空间和应用价值。传统的方法耗时长、设备昂贵、获取难...
基于单张rgb图像实现不同人体重建模型的融合方法.本发明涉及计算机视觉处理领域,尤其涉及一种基于单张rgb图像实现不同人体重建模型的融合方法。.人体三维重建是三维重建领域中的的重要细分领域。低成本、便捷的获取人体三维模型有广泛的应用空间和应用价值。传统的方法耗时长、设备昂贵、获取难... -
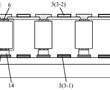
 .本发明涉及激光器,尤其涉及一种多级光纤放大器系统。.多级光纤放大是光纤激光器重要的环节,目前的多级光纤放大系统的光纤器件多,尾纤多,连接关系复杂,安装维护困难。.现有技术cna公开一种多级光纤放大器,包括盒体、第一隔板、第二隔板、多个光纤器件、第一支撑组件和第二支撑组件,所述第一...
.本发明涉及激光器,尤其涉及一种多级光纤放大器系统。.多级光纤放大是光纤激光器重要的环节,目前的多级光纤放大系统的光纤器件多,尾纤多,连接关系复杂,安装维护困难。.现有技术cna公开一种多级光纤放大器,包括盒体、第一隔板、第二隔板、多个光纤器件、第一支撑组件和第二支撑组件,所述第一... -
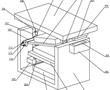
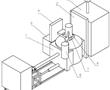 .本发明涉及精密铸造领域,特别涉及一种高温合金真空低压熔铸装置。.目前,国内高温合金精铸件一般采用真空感应熔炼、重力浇注的方法成形,该方法存在充型过程合金熔体填充形态可控性差,容易卷入非金属夹杂,对于壁厚差异大且具有大面积薄壁的复杂结构铸件,还容易产生浇不足和缩松缩孔等缺陷。低压铸造、真空...
.本发明涉及精密铸造领域,特别涉及一种高温合金真空低压熔铸装置。.目前,国内高温合金精铸件一般采用真空感应熔炼、重力浇注的方法成形,该方法存在充型过程合金熔体填充形态可控性差,容易卷入非金属夹杂,对于壁厚差异大且具有大面积薄壁的复杂结构铸件,还容易产生浇不足和缩松缩孔等缺陷。低压铸造、真空... -
 .本发明涉及路侧数据分析,具体涉及一种基于网联车的路侧数据交通分析方法及装置。.交通信息感知是交通信息基础设施的重要功能之一,为交通态势预测、信号控制等交通应用场景提供必要的数据和决策支持。目前常见的交通信息态势感知手段主要有地磁线圈、雷达、视频、红外等,但这些单一的交通信息感知设...
.本发明涉及路侧数据分析,具体涉及一种基于网联车的路侧数据交通分析方法及装置。.交通信息感知是交通信息基础设施的重要功能之一,为交通态势预测、信号控制等交通应用场景提供必要的数据和决策支持。目前常见的交通信息态势感知手段主要有地磁线圈、雷达、视频、红外等,但这些单一的交通信息感知设... -
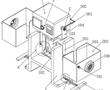 .本发明涉及无损检测的,尤其是涉及基于红外长脉冲的多重双波大型激光超声检测器。.激光超声已成为无损检测领域的一种成熟技术,因为它具有无需接触、高空间分辨率、亚纳米级灵敏度等优点且可用于复杂结构和高温环境,激光超声系统是传统压电换能器系统的更好替代选择,因为它无需耦合剂的,激光超声系...
.本发明涉及无损检测的,尤其是涉及基于红外长脉冲的多重双波大型激光超声检测器。.激光超声已成为无损检测领域的一种成熟技术,因为它具有无需接触、高空间分辨率、亚纳米级灵敏度等优点且可用于复杂结构和高温环境,激光超声系统是传统压电换能器系统的更好替代选择,因为它无需耦合剂的,激光超声系... -
.本发明涉及激光校靶,尤其是指一种基于激光校准用自定位目标靶装置。.激光校靶作为发射轴线与瞄准轴线一致性检测方式已被广泛使用,其工作原理:利用激光相干性和准直性好的特点,以激光作为发射轴线的理论延长线,激光光斑中心对准目标位置(发射轴线与靶板相交点),调整瞄准镜,使瞄准镜中心对准瞄...
-
.本发明涉及航空发动机高温合金定向/单晶铸件的熔模铸造,具体涉及一种定向/单晶高温合金铸件熔模铸造用惰性陶瓷型壳制备方法。.定向/单晶铸件熔模铸造浇注过程中,℃左右的高温合金液要在型壳中停留至少h,合金熔体中的活泼元素与型壳内腔表面易发生复杂的物理、化学相互作用,当物理、化学反应过...
-
 复合材料及其制备方法、电池.本申请涉及电池材料,尤其涉及一种复合材料及其制备方法、电池。.钛酸锂因具有高的嵌锂电位(.vvsli),不生成锂枝晶,并且充电和放电状态热稳定性高,作为锂离子电池的负极材料,具有优异的安全性。此外,充放电过程体积基本不发生变化,被称为“零应变...
复合材料及其制备方法、电池.本申请涉及电池材料,尤其涉及一种复合材料及其制备方法、电池。.钛酸锂因具有高的嵌锂电位(.vvsli),不生成锂枝晶,并且充电和放电状态热稳定性高,作为锂离子电池的负极材料,具有优异的安全性。此外,充放电过程体积基本不发生变化,被称为“零应变... -
.本发明涉及医用硅胶带,具体为一种医用硅胶胶带及其制备方法。.硅胶胶带是医院中常用的物品,但现有的硅胶胶带仍然存在不足之处,具体为:现有的硅胶胶带透气性较差,容易刺激皮肤,影响患者伤口愈合。.因此,需要一种医用硅胶胶带及其制备方法来解决上述中提出的问题。发明内容.本发明的目...
-
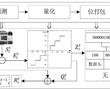 面向配电网边缘计算装置的d-pmu数据实时压缩方法.本发明涉及一种配电网同步相量测量单元(distribution-levelphasormeasurementunit,d-pmu)数据实时压缩方法。特别是涉及一种面向配电网边缘计算装置的d-pmu数据实时压缩方法。.各种先进...
面向配电网边缘计算装置的d-pmu数据实时压缩方法.本发明涉及一种配电网同步相量测量单元(distribution-levelphasormeasurementunit,d-pmu)数据实时压缩方法。特别是涉及一种面向配电网边缘计算装置的d-pmu数据实时压缩方法。.各种先进... -
 一种储层采油的炸药确定方法、系统、设备及介质.本发明涉及爆炸压裂,特别是涉及一种储层采油的炸药确定方法、系统、设备及介质。.目前已有一些理论研究储层的炸药爆裂改造,也取得了一定成效,但针对炸药对储层改造仍处于起步阶段,离实际应用还有很大差距。目前研究的重点仍局限于裂缝分形性...
一种储层采油的炸药确定方法、系统、设备及介质.本发明涉及爆炸压裂,特别是涉及一种储层采油的炸药确定方法、系统、设备及介质。.目前已有一些理论研究储层的炸药爆裂改造,也取得了一定成效,但针对炸药对储层改造仍处于起步阶段,离实际应用还有很大差距。目前研究的重点仍局限于裂缝分形性...
















利记sbobet网址
- 农业,林业,园林,畜牧业,肥料饲料的机械,工具制造及其应用技术
- 食品,饮料机械,设备的制造及其制品加工制作,储藏技术
- 烟草加工设备的制造及烟草加工技术
- 服装,鞋;帽,珠宝,饰品制造的工具及其制品制作技术
- 医药医疗技术的改进;医疗器械制造及应用技术
- 家居日用产品装置的制造及产品制作技术
- 休闲,运动,玩具,娱乐用品的装置及其制品制造技术
- 木材加工工具,设备的制造及其制品制作技术
- 纺织,织造,皮革制品制作工具,设备的制造及其制品技术处理方法
- 建筑材料工具的制造及其制品处理技术
- 家具;门窗制品及其配附件制造技术
- 水利;给水;排水工程装置的制造及其处理技术
- 道路,铁路或桥梁建设机械的制造及建造技术
- 五金工具产品及配附件制造技术
- 安全;消防;救生装置及其产品制造技术
- 造纸;纤维素;纸品设备的制造及其加工制造技术
- 印刷排版;打字模印装置的制造及其产品制作工艺
- 办公文教;装订;广告设备的制造及其产品制作工艺
- 工艺制品设备的制造及其制作,处理技术
- 摄影电影;光学设备的制造及其处理,应用技术
- 乐器;声学设备的制造及制作,分析技术
- 照明工业产品的制造及其应用技术
- 机械加工,机床金属加工设备的制造及其加工,应用技术
- 金属材料;冶金;铸造;磨削;抛光设备的制造及处理,应用技术
- 无机化学及其化合物制造及其合成,应用技术
- 有机化学装置的制造及其处理,应用技术
- 有机化合物处理,合成应用技术
- 喷涂装置;染料;涂料;抛光剂;天然树脂;黏合剂装置的制造及其制作,应用技术
- 车辆装置的制造及其改造技术
- 铁路车辆辅助装置的制造及其改造技术
- 自行车,非机动车装置制造技术
- 船舶设备制造技术
- 航空航天装置制造技术
- 包装,储藏,运输设备的制造及其应用技术
- 塑料加工应用技术
- 蒸汽制造应用技术
- 燃烧设备;加热装置的制造及其应用技术
- 供热;炉灶;通风;干燥设备的制造及其应用技术
- 制冷或冷却;气体的液化或固化装置的制造及其应用技术
- 环保节能,再生,污水处理设备的制造及其应用技术
- 物理化学装置的制造及其应用技术
- 分离筛选设备的制造及其应用技术
- 石油,煤气及炼焦工业设备的制造及其应用技术
- 发动机及配件附件的制造及其应用技术
- 微观装置的制造及其处理技术
- 电解或电泳工艺的制造及其应用技术
- 土层或岩石的钻进;采矿的设备制造及其应用技术
- 非变容式泵设备的制造及其应用技术
- 流体压力执行机构;一般液压技术和气动零部件的制造及其应用技术
- 工程元件,部件;绝热;紧固件装置的制造及其应用技术
- 气体或液体的贮存或分配装置的制造及其应用技术
- 测量装置的制造及其应用技术
- 测时;钟表制品的制造及其维修技术
- 控制;调节装置的制造及其应用技术
- 计算;推算;计数设备的制造及其应用技术
- 核算装置的制造及其应用技术
- 信号装置的制造及其应用技术
- 信息存储应用技术
- 电气元件制品的制造及其应用技术
- 发电;变电;配电装置的制造技术
- 电子电路装置的制造及其应用技术
- 电子通信装置的制造及其应用技术
- 其他产品的制造及其应用技术